
Синдром олбрайта как форма фиброзной дисплазии. Синдром Олбрайта: заболевание с необратимыми последствиями Что такое синдром олбрайта
Книга: «Редкие неврологические синдромы и болезни» (В.В. Пономарев)
Глава 2. Болезнь Олбрайта
Болезнь Олбрайта (синонимы: псевдогипопаратиреоз, синдром Мартина-Олбрайта, наследственная остеодистрофия Олбрайта, синдром «яванской курицы») - редкое наследственное заболевание, характеризующееся невосприимчивостью клеток-мишеней к паратгормону (ПТГ), которое впервые описано швейцарским врачом Е. Martin и его американским коллегой F. Albright. Болезнь наследуется по доминантному типу или Х-сцепленному с полом. В зависимости от места генетического дефекта выделяют два типа болезни Олбрайта (БО). При первом типе нарушена структура белков-рецепторов к ПТГ, при втором наблюдается дефект фосфор-кальциевых систем клеточных мембран. Клиническими и общедоступными лабораторными методами обследования дифференцировать оба типа невозможно, однако первый тип БО может входить в синдром множественной невосприимчивости рецепторов клеток-мишеней к гормонам (тиреотропину, гонадотропину и др.) .
Вследствие нарушения фосфор-кальциевого обмена внутри клеток (при этом показатели уровня электролитов крови могут быть в пределах нормы) страдают практически все органы и системы организма, что приводит к развитию специфической клинической картины. Ведущими клиническими синдромами БО являются низкий рост, изменение соотношения длины пальцев стоп, нарушение развития зубной ткани, увеличение толщины костей черепа. На поражение других тканей указывают кальцинация структур головного мозга, хрусталиков, мягких тканей и кожи, выпадение волос. Поражение нервной системы при БО проявляется эпилептическими приступами, фокальными тоническими судорогами в лице или конечностях, умственной отсталостью .
Мы наблюдали редкий семейный случай этой болезни.
Больной В., 16 лет, поступил с жалобами на головную боль в затылочной области, периодические приступы онемения и тонических судорог в конечностях (чаще в ногах), иногда с их генерализацией и кратковременной потерей сознания, прикусом языка. Из анамнеза: родился в срок доношенным. С раннего детства отставал от сверстников в физическом развитии. С 12 лет без видимой причины начали повторяться приступы тонических судорог в конечностях, иногда с их генерализацией (1-2 раза в месяц). Принимал противосудорожные средства (финлепсин) без эффекта. Из ранее перенесенных заболеваний отмечает менингоэнцефалит в возрасте 2 года, в 15-летнем возрасте оперирован по поводу врожденной катаракты обоих глаз. Объективно: маленького роста (145 см), со слабо развитой мышечной системой, ихтиоз кожи. Артериальное давление 90/60 мм рт. ст., пульс 72 уд/мин, ритмичный. Отмечаются множественные диспластические признаки костно-суставной системы: равномерное увеличение черепа в объеме, усилен грудной кифоз, сколиоз в грудном отделе, частичная контрактура плечевых, локтевых, коленных, тазобедренных суставов, гипермобильность межфаланговых суставов кистей, значительное удлинение фаланг II пальцев и частичная синдактилия II-III пальцев стоп. Снижена память, интеллект, критика к своему состоянию. Зрачки широкие, слабо реагируют на свет, язык со следами прикуса. Парезов конечностей, менингеальных знаков нет, координация не нарушена. В течение дня периодически появляются кратковременные судороги в кистях (типа «руки акушера») и стопах. Положительные симптомы Хвостека и Труссо.
При обследовании: общие анализы крови и мочи без патологии. Электролиты крови: кальций 2,0 ммоль/л (норма 2,0-2,7 ммоль/л), калий 3,4 ммоль/л (норма 3,6-5,4 ммоль/л), натрий 132 ммоль/л (норма 80-140 ммоль/л), фосфор 1,0 ммоль/л (норма 0,8-1,4 ммоль/л). СМЖ: белок 0,6 г/л, цитоз 45 106 клеток/л.
Острота зрения на оба глаза 0,01 с коррекцией 0,3, глазное дно в норме. ЭЭГ: умеренные диффузные изменения по типу дезорганизации коркового ритма. КТ головного мозга: значительное увеличение толщины костей свода черепа, множественные симметричные кальцинаты в лобных долях, проекции базальных ганглиев, белом веществе головного мозга и мозжечке (рис. 16). Назначен допакин (450 мг/сут), на фоне которого приступы тетании в руках уменьшились, судорожные приступы не повторялись.

Рис. 16. КТ головного мозга В., 16 лет, с диагнозом БО: значительное увеличение толщины костей свода черепа, кальцинаты в проекции базальных ганглиев
Активно осмотрены старший брат и мать больного. При обследовании брата К., 19 лет, установлено, что он также отставал с раннего детства в физическом развитии. В возрасте 1-7 лет наблюдались редкие (до нескольких раз в год) генерализованные судорожные приступы, которые в последующем не повторялись. При объективном осмотре выявлены аналогичные диспластические признаки патологии костно-суставной системы, выраженные в меньшей степени: низкий рост (153 см), ихтиоз кожи, увеличение объема черепа, легкая контрактура плечевых и тазобедренных суставов, гипермобильность V пальца правой кисти, удлинение II пальца и синдактилия II-III пальцев стоп. При офтальмоскопии выявлено помутнение хрусталиков, врожденная катаракта. КТ головного мозга: значительное утолщение костей свода черепа, кальцинация базальных ганглиев. Электролиты крови в норме. Со слов матери, оба брата внешне похожи на ее отца, который умер в возрасте 53 года и также страдал эпилептическими приступами. У матери признаков болезни не выявлено.
Таким образом, при объективном осмотре, лабораторном и инструментальном обследовании братьев В. (16 лет) и К. (19 лет) наблюдались схожие клинические признаки БО: множественная ахондроплазия, врожденная катаракта, трофические расстройства кожи (ихтиоз), деменция, приступы тетании, генерализованные приступы. В обоих случаях при нормальном содержании электролитов в крови на КТ головного мозга обнаруживалась массивная кальцификация. Установлен доминантный тип наследования болезни. Представляет интерес наличие судорожных приступов у брата В., которые имели вторично-генерализованный характер, начинаясь с явлений тетании, что ранее не описано в литературе.
Самый характерный признак БО - приступы тонических судорог в конечностях (тетания) - связан с повышенной возбудимостью нервно-мышечного аппарата. Достаточно часто в клинической картине заболевания встречаются эпилептические припадки (абсансы, лобно-долевые, генерализованные судорожные). В их основе лежат как нарушения нормального электрогенеза в нервной системе из-за кальциевого дисбаланса, так и непосредственное раздражение мозга кальцинатами .
Дифференциальный диагноз БО проводят с болезнью Фара, для которой не характерно наличие ахондроплазии, первичным или вторичным гипопаратиреозом.
Этиологической и патогенетической терапии БО к настоящему времени не существует. К числу симптоматических средств лечения этой патологии относится назначение противосудорожных средств (препаратов вальпроевой кислоты или карбамазепинов), витамина D по 100-200 тыс. ед.
Таким образом, БО имеет специфическую клиническую картину и некоторый положительный лечебный эффект может быть достигнут под влиянием симптоматической терапии.
Синдром Мак-Кьюна-Олбрайта-Брайцева – очень редкое заболевание, имеющее генетическую природу. Оно диагностируется лишь у одного человека из ста тысяч, чаще патология встречается у представительниц женского пола. Поражение сопровождается нарушением обмена кальция и фосфора, которое возникает на фоне нечувствительности рецепторов организма к воздействию паратгормона. Симптомы включают в себя костные деформации, отклонения функции эндокринных желез, отставание в умственном и физическом развитии. Лечение основано на использовании медикаментозных средств, в ряде случаев прибегают к проведению операций. Важное место в терапии занимает также особая диета и ограничение физических нагрузок.
Причины появления синдрома
Точная этиология недуга на сегодняшний день неизвестна. Ученые предполагают, что заболевание относится к числу генетических расстройств. При этом особенностью проблемы является ее неспособность передаваться по наследству. Мутации в хромосомах возникают спонтанно и провоцируют нарушения метаболизма, связанные со снижением чувствительности рецепторов почек и костной ткани к воздействию гормона паращитовидных желез. Подобный каскад реакций приводит к развитию гипопаратиреоза. Симптомы формируются вследствие сбоев в метаболизме кальция и фосфора. К числу провоцирующих факторов развития синдрома относят следующие негативные воздействия:
- Сильный стресс способен инициировать возникновение расстройства. Психологическое напряжение сказывается на работе желез внутренней секреции и приводит к формированию проблем.
- Врожденные аномалии развития гипоталамуса. Этот отдел головного мозга принимает активное участие в контроле функции эндокринной системы. Поэтому дефекты его строения способны приводить к развитию у человека синдрома Олбрайта.
- Различные сбои в работе желез внутренней секреции, например, изменение функции яичников, надпочечников и других структур. Поскольку все элементы эндокринной системы связаны между собой, сбои в работе одного звена способны нарушать естественный гомеостаз.
Основные симптомы генетического недуга
Клинические проявления болезни включают в себя:
- Деформации костей – фиброзная остеодисплазия. Возникновение подобных симптомов связано с нарушениями кальциево-фосфорного обмена. Возможны патологии строения позвоночника и конечностей. Это сопровождается развитием хромоты, боли и возникновением характерного внешнего вида пациента. Кости становятся хрупкими, что повышает риск переломов.
- Появление на коже пигментированных участков. Пятна могут иметь различный размер и локализацию. В ряде случаев дефекты стремительно растут и занимают значительную поверхность тела. Подобный симптом является основанием для проведения гистологического исследования пораженных участков кожи.
- Распространенный признак синдрома Олбрайта – судороги. Они также связаны с нарушением обмена кальция, который принимает участие в работе нервной системы. Патологическая активность мышц часто провоцируется воздействием неблагоприятных факторов, например, стресса.
- Пациенты с заболеванием страдают от гормональных расстройств. У девочек отмечается раннее половое созревание – появление менструации и увеличение размеров молочных желез.
- Характерна для синдрома Мак-Кьюна-Олбрайта-Брайцева задержка развития, как умственного, так и физического. Пациенты с недугом не выглядят на свой возраст и зачастую нуждаются в занятиях с дефектологом.
- Люди с заболеванием имеют характерный внешний вид. Пациенты невысокого роста, зачастую страдают от ожирения, у них лунообразное лицо и широко посаженные глаза. Фото пациентов представлены ниже.
К числу редких осложнений синдрома Олбрайта относят формирование злокачественных новообразований. Их возникновение в большинстве случаев связано с перерождением участков фиброзной дисплазии костных структур, которая является одной из характерных особенностей заболевания. Подобное последствие патологии диагностируется лишь у 1% пациентов. Среди факторов, провоцирующих появление данной проблемы – инсоляции, то есть регулярное пребывание на солнце, что связано с воздействием ультрафиолетовых лучей.
Повышен риск развития опухолевого процесса и у пациентов с увеличением концентрации гормона роста в крови. Самым распространенным вариантом онкологии считаются остеосаркомы, характеризующиеся высокой устойчивостью к лечению, поэтому имеющие осторожный или неблагоприятный прогноз. Среди доброкачественных проблем, возникающих при синдроме Олбрайта, ведущее место занимают внутримышечные миксомы, в случае обнаружения которых рекомендуется осуществление хирургического вмешательства.
Увеличивается также риск заболевания раком молочных желез. У всех пациентов с этим поражением отмечалось значительное завышение уровня гормона роста, что дает основание предполагать роль соединения в развитии онкологии.
Проведение диагностики
Зачастую заподозрить синдром Олбрайта сразу после рождения ребенка не удается. Однако по мере его роста и развития происходит возникновение типичной клинической картины. При наличии деформаций костей, пигментных пятен и эндокринопатий постановка диагноза не вызывает трудностей у врачей. Для оценки степени выраженности изменений используется рентген. Специфическим является тест с определением количества фосфатов в моче после стимулирующего введения паратгормона. Используются и гематологические тесты. Поставить точный диагноз можно по результатам генетического анализа.
Методы лечения
Тактика борьбы с заболеванием индивидуальна в каждом случае. Она зависит от степени выраженности симптомов. Используется как консервативная терапия, так и хирургические техники. Важным этапом лечения синдрома Олбрайта является также сбалансированное питание и выполнение упражнений, направленных на укрепление мышечного аппарата. Следует учитывать, что борьба с недугом носит симптоматический характер. Она направлена на снижение выраженности последствий эндокринных нарушений. Излечить заболевание полностью не представляется возможным.

Использование медикаментов
Для коррекции состояния пациентов назначают следующие средства:
- Основу терапии составляет применение препаратов кальция. Они используются в довольно высоких дозировках, достаточных для поддержания физиологического уровня элемента в организме пациента. Это необходимо для предупреждения развития излишней хрупкости костей и профилактики прогрессирования умственной отсталости.
- Прием витамина D также важен для восстановления нормального обмена кальция и фосфора. Он назначается в сочетании с приемом микроэлементов, например, используются комбинированные средства, такие как «Натекаль Д3». Это соединение также играет важную роль в поддержании функции эндокринной системы и здоровья кожи.
- Антиандрогены, к числу которых относится препарат «Финастерид», применяются при лечении синдрома Олбрайта для предупреждения преждевременного полового созревания у девочек.
Хирургическое вмешательство
К радикальным тактикам прибегают в случае угрозы повреждения нервных структур, например, при ухудшении зрения вследствие деформации черепа. Проводятся операции и при переломах, сопровождающихся смещением костей. Хирургическое вмешательство зачастую также требуется больным с затрудненным дыханием. Если пациент страдает от формирования камней в почках или состояний, сопровождающихся нарушением оттока мочи, проводятся соответствующие операции.
Народные способы
Лечиться можно и в домашних условиях, но только с разрешения врача.
- Источником кальция является скорлупа куриных или перепелиных яиц. Их предварительно хорошо моют и очищают от загрязнений. Ингредиент сушат и мелко перемалывают до состояния порошка. Скорлупу можно смешать с небольшим количеством лимонного сока. Средство принимают по половине чайной ложки три раза в день на протяжении двух недель.
- Антиандрогенным эффектом обладает сочетание экстрактов стевии и карликовой пальмы. Эти средства можно приобрести в аптеках и принимать по инструкции для предотвращения раннего полового развития у девочек.
Правильное питание и добавки
Пациентам требуется диета с повышенным содержанием кальция и низкой концентрацией фосфора. Это предполагает присутствие в рационе таких продуктов, как бобы, чечевица, молоко и укроп. Ограничивается потребление мяса, яиц, сыра и рыбы.
Возможно и применение кальция в виде добавок к пище. В таких случаях препараты принимаются курсом после консультации с врачом.
Необходимые упражнения
Серьезные физические нагрузки пациентам противопоказаны. Вся активность должна быть направлена на общее укрепление мышц. С этой целью больным рекомендуются пешие прогулки, плавание, езда на велосипеде. Можно выполнять «мельницу», повороты корпуса, «ножницы» и приседания. Все занятия начинают с разминки. Рекомендуется заниматься физкультурой под присмотром опытного тренера.
Профилактика и прогноз
Специфических способов предупреждения развития синдрома Олбрайта нет. Поэтому недопущение формирования патологии сводится к соблюдению принципов здорового образа жизни.
Прогноз при своевременном лечении благоприятный при условии отсутствия сопутствующих проблем. Известным примером человека с синдромом Олбрайта, который, несмотря на заболевание, ведет весьма активный образ жизни, является Лилия Лис. Девушка своим примером мотивирует многих людей с различными проблемами со здоровьем.
УДК616.71-007.1-056.7: -036.1 ОО!: 10.22141/2224-0713.3.89.2017.104252
Пономарев В.В., Карасев Ю.А.
Белорусская медицинская академия последипломного образования, г. Минск, Беларусь
Болезнь Олбрайта: клинический случай и анализ литературы
Резюме. Представлена диагностика орфанного двигательного расстройства - спорадического случая болезни Олбрайта. Неврологические нарушения у пациента включали умеренное когнитивное снижение подкоркового типа, хореоатетоидный гиперкинез в левых конечностях и легкий экстрапирамидный синдром. Диагноз подтвержден лабораторными данными (высоким уровнем паратгормона, низким уровнем кальция), остеоденситометрией (остеопения шейки бедренной кости слева), рентгенографией коленных суставов (множественные мелкие обызвествления сливного характера в мягких тканях параартикулярно), характерными признаками при проведении компьютерной томографии головного мозга. Ключевые слова: болезнь Олбрайта; диагностика; лечение
М1ЖНАРОДНИЙ НЕВРОПОГ1ЧНИЙ ЖУРНАЛ
INTERNATIONAL NEUROLOGICAL JOURNAL |
МЕЖДУНАРОДНЫЙ НЕВРОЛОГИЧЕСКИЙ ЖУРНАЛ МАТЕР1АЛИ КОНФЕРЕНЦП /PROCEEDINGS OF THE CONFERENCE/
Болезнь Олбрайта (БО, наследственная остеоди-строфия, псевдогипопаратиреоз) - редкое наследственное полисистемное заболевание, характеризующееся невосприимчивостью клеток-мишеней к па-ратгормону . Его истинная частота неизвестна, однако считается, что БО относится к числу орфан-ных болезней, так как в мировой литературе к концу XX века было описано только около 300 случаев . По механизму развития БО напоминает гипопарати-реоз, в основе которого лежит первичный дефицит паратиреоидного гормона, обусловленный снижением функциональной активности паращитовидных желез. При БО, напротив, клинические проявления вызываются нарушением чувствительности тканей-мишеней к действию паратгормона при его достаточном уровне секреции. Впервые эта патология описана в 1942 г. Ф. Олбрайтом (F. Albright) и в последующем получила эпонимический термин . Заболевание обычно проявляется у детей и подростков .
Этиология БО неизвестна, хотя считается, что это генетически гетерогенное заболевание. Однако данные о типе ее наследственной передачи в литературе противоречивы. При БО описываются X-сцепленный, аутосомно-доминантный и аутосомно-рецессивный типы . В большинстве случаев развитие
БО связано с мутациями в расположенном на хромосоме 20 локусе 20q13 гена GNAS1. Наиболее часто наблюдается интерстициальная делеция длинного плеча второй хромосомы в локусе 2q37 . Изучение родословных показывает, что число женщин с БО в 2 раза превышает количество мужчин, так как установлено, что БО не передается от отца к сыновьям . В основе патогенеза БО лежит генетически обусловленная резистентность почек и костей к действию паратгор-мона в результате дефекта комплекса «специфический циторецептор - паратгормон - аденилатциклаза», что нарушает процесс образования в почках циклического 3",5"-АМФ, являющегося внутриклеточным посредником действия паратгормона на метаболические процессы .
В зависимости от биохимического уровня поражения выделяют несколько типов БО .
1-й А тип: дефектен сам циторецептор, связывающий паратгормон. Имеет аутосомно-доминантный тип наследования. При данном типе нередко наблюдается одновременное вовлечение многих эндокринных желез: щитовидной и поджелудочной желез, гонад.
1-й В тип: отмечается дефект нуклеотидсвязыва-ющего белка, локализованного в липидном слое клеточной мембраны, который функционально связывает
© «Международный неврологический журнал», 2017 © «International Neurological Journal», 2017
© Издатель Заславский А.Ю., 2017 © Publisher Zaslavsky O.Yu., 2017
Для корреспонденции: Пономарев Владимир Владимирович, Государственное учреждение образования «Белорусская медицинская академия последипломного образования», ул. П. Бровки, 3, г. Минск, 220013, Беларусь; e-mail: [email protected] For correspondence: Vladymir Ponomarev, State Education Institution"Belarusian Medical Academy for Postgraduate Education", P. Brovki st., 3, Minsk, 220013, Belarus; e-mail: [email protected]
рецептор с аденилатциклазой. Также имеет аутосом-но-доминантный тип наследования, однако не исключен сцепленный с Х-хромосомой тип. В отличие от 1-го А типа для данного типа нехарактерно поражение эндокринных желез.
2-й тип обусловлен ферментативной недостаточностью аденилатциклазы. Паратгормон при этом связывается с рецепторами и вызывает нормальную ответную реакцию клеток на паратгормон в виде увеличения экскреции циклического аденозинмонофосфата (цАМФ). Однако внутриклеточная нечувствительность к цАМФ не позволяет в полной мере реализоваться действию па-ратгормона. Отличие данного типа от остальных форм БО заключается в том, что сохраняется нормальная реакция почек на экзогенное введение паратиреоидного гормона в виде увеличения экскреции цАМФ с мочой.
Некоторые авторы выделяют БО, которая характеризуется отсутствием гипокальциемии, гиперфос-фатемии, судорог и остеомаляции, и относят ее к 1-му С типу. Однако вероятно, что этот вариант является одним из клинических фенотипов 1-го А типа .
Клиническая картина БО отличается мультиси-стемным и мультиорганным поражением, при котором соматические симптомы комбинируются с признаками поражения нервной системы . У пациентов отмечаются диспропорциональность физического развития, низкий рост (до карликовости) за счет укорочения нижних конечностей, резкое укорочение I, III и V пястных и плюсневых костей (особенно III и IV), «лунообразное» лицо . Вследствие нарушений минерального обмена иногда наблюдаются экзостозы и аплазия зубов; компенсаторная гиперплазия пара-щитовидных желез (наличие в них аденом не характерно); кальцификация мягких тканей, подкожные кальцификаты; может отмечаться остеопороз; поражение глаз (лентикулярная катаракта); кальцинаты в подкожной клетчатке с тенденцией к их изъязвлению, что имитирует оссифицирующий миозит. В части случаев при БО наблюдается одновременное вовлечение эндокринных желез, что проявляется гипотиреозом, гипо-гонадизмом, сахарным диабетом, усугубляет течение БО и придает его клиническим проявлениям выраженный полиморфизм .
Неврологические проявления считаются ведущими нарушениями при БО, так как обусловлены симметричной интрацеребральной кальцификацией коры полушарий, базальных ганглиев и зубчатых ядер мозжечка в связи с отложением солей кальция и железа в стенках мелких артерий и артериол, а также в веществе головного мозга . Выделяют несколько ведущих клинических синдромов, которые у пациентов с БО встречаются в различных комбинациях и разной степени выраженности. К ним относят: 1) подкорковую деменцию; 2) локальные судороги и/или тетанические спазмы; 3) экстрапирамидные нарушения: гиперкине-зы (хорея, тремор, дистония, атетоз, орофациальная дискинезия) или паркинсонизм; 4) мозжечковые симптомы; 5) нарушение речи; 6) преходящие или стойкие пирамидные знаки; 7) эпилептические приступы;
8) боль в дистальных отделах конечностей; 9) положительные симптомы Хвостека и Труссо .
К основным диагностическим критериям БО относят :
Низкий рост;
Круглое лицо;
Задержку нервно-психического развития;
Скелетные аномалии;
Высокий уровень паратиреоидного гормона в крови;
Снижение экскреции с мочой фосфатов и цАМФ.
Диагностика БО помимо характерной клинической
картины основывается на лабораторных данных. Дифференциально-диагностическим тестом может быть характер почечной экскреции цАМФ в ответ на введение паратгормона: повышенная экскреция цАМФ отмечается при типе 2 и ее отсутствие - при типе 1 . Диагноз БО подтверждается обнаружением сниженного уровня гуаниннуклеотидсвязывающего белка белок) в крови (в среднем в 1,5-2 раза) по сравнению с нормой . Гипокальциемия, как правило, сочетается с гиперфосфатемией и гипофосфатурией. Уровень паратгормона повышен, однако при 1С-типе его уровень может быть в норме . При рентгенологическом исследовании костной системы у части пациентов с БО обнаруживается укорочение пястных и плюсневых костей, нередко - генерализованная деминерализация (остеопороз), значительное утолщение костей свода черепа .
Лечение БО заключается в назначении препаратов кальция в дозах, достаточных для поддержания нормальной концентрации кальция в крови . Большое значение имеет терапия витамином D. Начальную дозу рассчитывают из 2000 МЕ/кг массы тела в сутки, но не более 100 000 МЕ в сутки. Во избежание передозировки препаратов витамина D необходим контроль за концентрацией кальция в крови каждые 3-7 дней в течение первых двух недель лечения и каждый месяц в течение последующих 2-3 месяцев. По достижении стабильной концентрации кальция в крови достаточно проверять ее 1 раз в 2-3 месяца . Можно применять кальцитрин, дигидротахистерол, оксидевит, а также другие препараты активных форм витамина D. Важное значение придают диете с ограничением фосфора, что помогает нормализовать концентрацию кальция в крови и устранить симптомы вторичного гиперпа-ратиреоза . При недостаточности других желез внутренней секреции проводят заместительную коррекцию соответствующими гормонами. При неврологических нарушениях используют симптоматическую
терапию: противосудорожные препараты; антиокси-данты, мозговые метаболиты; препараты леводопы; клоназепам .
Прогноз при БО точно не определен, однако можно предположить, что своевременная диагностика и рациональная терапия этой патологии позволяют говорить о положительных прогнозах на жизнь и возможности контроля за течением заболевания .
Ранее (в 2005 году) мы уже приводили семейный случай диагностики БО , однако исключительная редкость описания спорадических случаев этой патологии в русскоязычной литературе побудила нас привести еще одно собственное клиническое наблюдение, при этом сопроводив его анализом современной литературы.
Пациент Г., 36 лет, холост, на момент обращения не работает, инвалид 3-й группы, образование среднее специальное. Жалобы при поступлении на насильственные движения в левых конечностях, больше в руке; онемение в левой руке, нарушение речи; плохой сон. Считает себя больным с июня 2001 г., когда появились периодические тонические сокращения мышц правой половины лица с присоединением насильственных вычурных движений в правых конечностях продолжительностью 10-15 секунд 5-6 раз в сутки, возникающие после провоцирующего фактора (перемена положения тела, физическая нагрузка, внезапное речевое обращение к пациенту). 26.11.2001 г. был госпитализирован в одно из неврологических отделений в г. Минске, где проведена компьютерная томография головного мозга и на основании заключения специалиста по лучевой диагностике установлен диагноз «болезнь Фара». При выписке была рекомендована постоянная противосу-дорожная терапия карбамазепином. К 2006 г. на фоне регулярного приема данного препарата двигательные пароксизмы регрессировали, однако появились и начали усиливаться когнитивные нарушения. За последние несколько лет появились насильственные движения в левых конечностях, нарастала выраженность когнитивных нарушений, присоединилось нарушение речи. В течение всего периода заболевания пациент неоднократно проходил лечение в различных неврологических отделениях г. Минска с диагнозом «болезнь Фара». Анамнез жизни: отмечается патология перинатального периода: осложненные роды у матери, у новорожденного выявлен врожденный порок сердца (недостаточность митрального клапана). Отмечался логоневроз с детства, начал говорить только с 4 лет. Семейно-на-следственный анамнез достоверно не отягощен, однако мать также имеет низкий рост и множественные стигмы строения скелета. В 1996 г. оперирован по поводу врожденного косоглазия. С 2003 по 2006 г. проходил лечение у офтальмологов с диагнозами: нейроретинит О8, кератопатия О8, артифакия, незрелая, осложненная катаракта ОD; оперировали катаракту О8 (2005 г.).
При объективном осмотре: общее состояние удовлетворительное, выглядит моложе своих лет, телосложение нормостеническое, алиментарное ожирение. Костно-мышечная система: низкий рост, равновели-
кая длина пальцев стоп, размер черепа непропорционален скелету, короткие широкие фаланги пальцев. АД 120/80 мм рт.ст., границы сердца не расширены, тоны сердца ясные, ритмичные; дыхание везикулярное. Язык влажный, не обложен, живот мягкий, безболезненный, печень не увеличена, мочеиспускание, стул - без особенностей, симптом Пастернацкого отрицательный. Неврологический статус: в сознании, эйфоричен, снижена память, ослаблено внимание, элементы бра-дифрении. Глазодвигательных нарушений, нистагма нет. Лицо симметрично. Язык по средней линии. Речь умеренно дизартрична, доступна пониманию. Объем активных и пассивных движений в конечностях сохранен. Несколько повышен тонус в левых конечностях, по экстрапирамидному типу - больше в руке. Глубокие рефлексы равномерно оживлены, D = 8. Силовых парезов не выявлено. Почти постоянный среднеампли-тудный неритмичный хореоатетоидный гиперкинез в левых конечностях, больше выражен в руке. В позе Ромберга устойчив. Координаторные пробы выполняет удовлетворительно. Нейропсихологическое тестирование: ММ8Е - 26 баллов, БАБ - 8 баллов (характерны для умеренного подкоркового типа когнитивного снижения).
Лабораторные исследования. Общеклинические анализы крови и мочи - без особенностей. Биохимический анализ крови: кальций - 1,32 ммоль/л (норма 2,2- 2,65 ммоль/л); фосфор - 2,03 ммоль/л (норма 0,81- 1,45 ммоль/л). Гормональное исследование крови: АТ-ТПО - 63,06 мМЕ/мл (норма 0-34 мМЕ/мл), Т3св. - 2,96 пм/л (норма 3,9-6,0 пм/л), Т4св. - 8,89 пм/л (норма 12-22 пм/л), остеокальцин - 43,55 пг/мл (норма 24-70 пм/л), паратгормон - 348,3 пг/мл (норма 15-65 пм/л), ТСГ - 5,78 мМЕ/мл (норма 0,5-4,67 мМЕ/мл), кальций ионизированный - 0,32 ммоль/л (норма 1,0-1,2 ммоль/л). Эти результаты указывали на гипофункцию щитовидной железы с аутоиммунным компонентом. Содержание паратгормо-на в несколько раз превышало норму. Уровень кальция в крови (общий и ионизированный) был ниже нормальных величин, а фосфора - напротив, повышен. Данный дисбаланс является патогномоничным для БО.
Инструментальные исследования. Сцинтиграфия паращитовидных желез: данных в пользу гиперфункциональной гормонально активной аденомы паращи-товидных желез не выявлено. Остеоденситометрия поясничного отдела позвоночника и шеек бедренных костей: незначительная остеопения шейки бедренной кости слева. УЗИ щитовидной железы: диффузная гиперплазия. Рентгенография коленных суставов: рентген-признаки гонартроза 1-2-й ст., множественные мелкие очаговые тени (обызвествления), местами сливного характера, в мягких тканях параартикулярно. ЭЭГ: умеренные изменения ЭЭГ дизритмичного характера с признаками дисфункции срединно-стволовых структур головного мозга (8 > D), патологических форм активности, очаговых нарушений нет. КТ головного мозга: линейные участки обызвествления в базальных ганглиях; пятнистые участки обызвествления белого
Рисунок 1. КТголовного мозга пациента Г., 36лет, с диагнозом «болезнь Олбрайта»: отмечается значительное утолщение костей свода черепа, пятнистые участки обызвествления белого вещества в лобных, теменных, затылочных и височных долях головного мозга
вещества в лобных, теменных, затылочных и височных долях головного мозга (рис. 1), обоих полушариях мозжечка и проекции ствола (рис. 2), утолщены кости свода черепа (рис. 1).
Осмотры специалистов. Уролог: без острой урологической патологии; эндокринолог: рекомендовано исследование кальция общего и ионизированного, уровня паратгормона; психиатр: когнитивно-мнестически снижен, эмоциональная лабильность в структуре органической патологии центральной нервной системы; логопед: дизартрия.
Учитывая характер жалоб, данные анамнеза, соматического, неврологического статуса, результаты лабораторных и инструментальных методов иссле-
Рисунок 2. КТ головного мозга этого же пациента: пятнистые участки обызвествления белого вещества в обоих полушариях мозжечка и в проекции ствола
дования, пациенту был установлен диагноз: болезнь Олбрайта с умеренным хореоатетоидным гиперкинезом преимущественно в левой руке, когнитивными нарушениями подкоркового типа, псевдогипопарати-реозом. Пациенту назначены витамин D, препараты кальция, акатинол мемантин, топирамат, строгая диета. При осмотре в дальнейшем отмечена некоторая положительная динамика в отношении выраженности хореоатетоидного гиперкинеза.
Мы проводили дифференциальную диагностику со всеми заболеваниями, упомянутыми выше, которые могут сопровождаться схожими клиническими и рентгенологическими данными. Наибольшую сложность вызвала дифференциальная диагностика с болезнью Фара, по поводу которой пациент наблюдался в течение длительного времени. В табл. 1 представлены отличия
Болезнь Олбрайта
Болезнь Фара
Дизрафические признаки
Наследственный характер
Паратгормон
Норма или снижен
Сопутствующая резистентность к другим гормонам
Половые гормоны, глюкагон, АДГ
Опорно-двигательная система
Остеопороз
Может быть остеосклероз
Неврологические проявления
Чаще гиперкинезы
Чаще паркинсонизм
КТ головного мозга
Стриопаллидозубчатый кальциноз + гиперостоз свода черепа
Стриопаллидозубчатый кальциноз
Проба с паратгормоном
Отрицательна в большинстве случаев
Таблица 1. Дифференциальная диагностика болезни Фара и Олбрайта
между этими заболеваниями, анализ которых позволил нам остановиться на диагнозе БО.
Из таблицы следует, что в пользу БО указывали дизрафические признаки пациента (низкий рост, равновеликая длина пальцев стоп, размер черепа непропорционален скелету, короткие широкие фаланги пальцев), уровень паратгормона, значительно превышающий нормальные показатели; гипофункция щитовидной железы с аутоиммунным компонентом; начальные стадии остеопороза; превалирование в неврологическом статусе гиперкинетического синдрома; на компьютерной томографии головного мозга, помимо стриопаллидозубчатого кальциноза, отмечался также гиперостоз свода черепа (толщина костей черепа в среднем на 2,5 мм превышала средние величины); проба с паратгормоном не привела к каким-либо значительным изменениям уровня кальция в сыворотке крови.
Интерес к данному клиническому случаю обусловлен исключительной редкостью заболевания, сложностью дифференциальной диагностики и полиморфизмом его клинических проявлений. Несмотря на распространенность поражения вещества головного мозга по результатам нейровизуализации, в неврологическом статусе у пациента отсутствовали мозжечковые и пирамидные симптомы. Клинический диагноз в нашем случае требует дополнительной ДНК-диагностики для его окончательной верификации и оценки риска появления БО у потомства.
Список литературы
1. Пономарев В.В. Редкие неврологические синдромы и болезни. - СПб.: Фолиант, 2005. - С. 105-109.
2. Лихачев С.А., Дрозд И.С., Корбут Т.В. Наследственная остеодистрофия Олбрайта (псевдогипопаратиреоз типа 1 а) с кальцификацией базальных ганглиев // Невролог. журнал. - 2007. - № 5. - С. 21-25.
3. Казанцева Л.З., Новиков П.В., Белова Н.А. и др. Наследственная остеодистрофия Олбрайта (псевдогипопаратиреоз) у детей//Рос. вестн. перинатол. и педиатр. - 1998. - № 5. - С. 43-45.
4. Пономарев В.В., Науменко Д.В. Болезнь Фара: Клиническая картина и подходы к лечению // Журнал невропатологии и психиатрии. - 2004. - № 3. - С. 62-64.
5. Пономарев В.В. Нейродегенеративные заболевания: настоящее и будущее // Мед. новости. - 2007. - № 5. - С. 23-28.
6. Albright F., Burnett C.H., Smith P.H., Parsons W. Pseudopara-thyroidism - an example of the Seabright-Bantam syndrome. Report of3 cases // Endocrinology. - 1942. - 30. - 922-935.
7. Ringel M.D., Schwinger W.E., Levine M.A. Clinical implications of genetic in G protein // Medicine (Baltimore). - 1996. - 75. - 4. - 171-184.
8. Phelan M.C., Rogers R.C., Clarkson K.B. et al. Albright hereditary osteodystrophy and del (2) (q37.3) in four unrelated individuals // Am. J. Med. Genet. - 1995. - 58. - 1. - 1-7.
9. Spiegel A.M. The molecular basis ofdisorders caused by defects in Gproteins// Horm. Res. - 1997. - 47. - 3. - 89-96.
10. Wilson L.C., Leverton K., Oude L.M.E. et al. Brachydactily and mental retardation: an Albright hereditary osteodystrophy-like syndrome located to 2q37// Am. J. Med. Genet. - 1995. - 56. - 2. - 400-407.
11. Koo B.B., Schwindiger W.F., Levine M.A. Characterization of Albright hereditary osteodystrophy and related disorders // Acta. Pediatr. Scan. - 1995. - 36. - 1. - 3-13.
12. Konupcikova K., Masopust J. Dementia in a patient with Fahr"s syndrome // Neuroendocrinology. - 2008. - №4. - Р. 431-434.
Получено 27.02.2017 ■
Пономарьов В.В., Карасьов Ю.О.
Блоруська медична академя пюлядипломно!" освпи, м. Мнськ, Блорусь
Хвороба Олбрайта: кл^чний
Резюме. Подана дiагностика орфанного рухового розладу - спорадичного випадку хвороби Олбрайта. Невролопчш по-рушення в пащента включали помiрне когштивне зниження пщюркового типу, хореоатетощний гшерюнез в лiвих юнщв-ках i легкий екстрашрамщний синдром. Дiагноз пщтвердже-ний лабораторними даними (високим рiвнем паратгормона,
випадок i ohoaî3 л^ератури
низьким piBHeM кальцш), остеоденситометрieю (остеопетя шийки стегново! шстки злiва), peнтгeногpафieю колшних cyrao6iB (множинт,npi6Hi звапншня зливного характеру в м"яких тканинах параартикулярно), характерними ознака-ми при проведент комп"ютерно! томографи головного мозку. слова: хвороба Олбрайта; дiагностика; лшування
V.V. Ponomariov, Yu.A. Karasev
Belorussian Medical Academy of Postgraduate Education, Minsk, Belorus
Albright Disease: A Clinical
Abstract. The paper deals with the case of orphan motor disorders - sporadic Albright osteodystrophy. Neurological signs in this case include moderate subcortical mental retardation, choreoath-etosis in left arm and mild extrapyramidal symptom.The correct diagnosis was verified by specific laboratory (high parathyroid hor-
Case and Literature Analysis
mone level, low concentrations of calcium) osteodensimetry indices (femoral neck left-side osteopenia), knee radiograms (multiple little confluent calcification in soft tissues para-articular) and CKT typical findings.
Keywords: sporadic Albright disease; diagnostic; therapy
Синдром Олбрайта – это наследственная патология с неясным происхождением. Ее еще называют псевдогипопаратиреозом. Для него характерно поражение костной ткани. Заболевание проявляется в основном у женской половины населения, хотя встречается и у мальчиков. Практически у всех больных отмечаются отклонения в умственном и физическом развитии. Кости человека начинают разрушаться под влиянием обменных процессов фосфора и кальция. Начинается выделение паратгормона паращитовидной железой.
- Этиология
- Симптоматика
- Диагностика
- Лечение
- Профилактика
Этиология
Основная причина такой патологии, как синдром Форбса – Олбрайта – это наследственная устойчивость тканей организма к влиянию паратгормона. Если человек не болен, тогда гормон действует посредством определенного вещества. Когда заболевание проявляется, то состав этого веществ изменяется на генетическом уровне. Таким образом периферические ткани становятся нечувствительными к паратгормону.
Симптоматика
Синдром Олбрайта имеет три основных симптома:
- Из-за высокого гормонального уровня происходит раннее половое созревание. Месячные у девочек могут начаться в период первых десяти месяцев жизни. Но этот симптом не относится к мальчикам. У них половое созревание начинается как обычно. Гормональный фон увеличивается, когда происходят сбои в работе надпочечников, щитовидной железы и гипофиза. Развиваться этот симптом может в двух формах ‒ неполной и полной. Если патология полной формы, тогда месячные начинаются слишком рано. При этом они болезненные и обильные. Также появляются и вторичные половые признаки, например, увеличиваются молочные железы. Если форма патологии ‒ неполная, тогда месячные не начинаются. Часто болезнь Олбрайта у девочек сопровождается развитием кисты на яичниках. Раннее половое созревание мальчиков характеризуется увеличением полового органа и появлением волос на лобке.
- Фиброзная остеодисплазия. Это патология, при которой деформируется костная ткань, из-за чего искривляются кости скелета. Проявляется в виде асимметричности. Визуальные симптомы ‒ это искривление позвоночника и хромота. Такая патология поражает в основном трубчатые кости. Рост человека замедляется. Это можно заметить даже у ребенка. Неравномерно развиваются кости черепа, появляется пучеглазие. После того как закончится половое созревание, изменение костей приостановится.
- Изменение цвета кожи. Начинает появляться сильная пигментация. На бедрах и самом теле проступают желто-коричневые пятна с нечеткими краями. Пятна хорошо видны на пояснице, верхней части спины, груди и ягодицах. При синдроме Олбрайта пятна на теле ребенка появляются сразу после его рождения.
Заболевание псевдогипопаратиреоз проявляет симптомы специфического характера.
Диагностика
При заболевании псевдогипопаратиреозом проводится диагностика, основанная на визуальном осмотре.
Также применяются лабораторные и аппаратные исследования:
- диагноз ставят при появлении двух основных симптомов;
- назначается консультация генетика;
- проводится специальный рентген;
- проводят лабораторный анализ крови на гормоны;
- требуется консультация детского эндокринолога;
- девочек осматривает детский гинеколог, а мальчиков ‒ уролог;
- девочкам назначают ультразвуковое обследование органов нижнего таза, а мальчикам проводят УЗИ мошонки;
- при необходимости назначают дополнительные обследования эндокринной системы.
Больные, страдающие от синдрома Олбрайта, должны регулярно проходить осмотры у таких специалистов, как эндокринолог, окулист, гинеколог, травматолог.
Лечение
Псевдогипопаратиреоз лечится комплексно.
Так как это генетическая патология, то терапия будет направлена на устранение появившихся симптомов:
- Назначается гормональное лечение. Это поможет в работе эндокринной системы. Необходимо внимательно следить за деформацией костей черепа, чтобы вовремя предотвратить развитие патологии.
- Оперативное вмешательство. Такой способ применяют, когда деформация лицевых костей может привести к нарушению слуха и зрения.
- Назначают обезболивающие препараты с содержанием бисфосфонатов.
Для каждого больного, страдающего от синдрома Олбрайта, способ терапии подбирается в индивидуальном порядке. Пациент должен строго соблюдать все рекомендации и назначения врача.
Проводится периодический контроль уровня кальция в организме пациента. На начальном этапе лечения такое исследование проводится один раз в неделю. Постепенно частота замеров снижается до одного раза в месяц. Она будет проводиться до конца назначенного лечения.
Если диагностирован псевдогипопаратиреоз, тогда больному необходимо придерживаться строгой диеты. Она заключается в полном исключении из меню продуктов, в которых присутствует фосфор.
Олбрайта синдром (F. Albright) - это заболевание, характеризующееся триадой признаков: фиброзная остеодисплазия, очаги кожной гиперпигментации и преждевременное половое созревание. Значительно чаще встречается у женщин.
Этиология синдрома Олбрайта неизвестна; предполагают неврогенные влияния и эндокринные сдвиги, вызванные врожденными изменениями в гипоталамической области, с преждевременной продукцией гонадотропного гормона передней доли гипофиза на почве склероза и гиперостоза основания черепа» главным образом в области турецкого седла (рис.).
Рост и зрелость скелета (появление и слияние ядер окостенения) могут значительно опережать нормальные показатели. Вследствие этого носители синдрома Олбрайта в детстве могут быть выше, а после преждевременного слияния эпифизов - ниже здоровых сверстников. Опережению развития скелета сопутствует преждевременное половое созревание - иногда стертое, редко значительное, когда менструальная функция может наступить в первые годы, а спорадические меноррагии - даже в первые месяцы жизни. Преждевременная половая зрелость у мальчиков редка.
Очаги гиперпигментации кожи при синдроме Олбрайта наблюдаются на туловище, бедрах, реже на шее, голенях. Они имеют вид крупных полей или отдельных и сливающихся пятен диаметром от 2 до 20 см и более, неправильной формы с изрезанными краями или представлены скоплениями веснушек от бледно-желтого до коричневого цвета. Иногда эти очаги видны уже при рождении, но при малой величине и бледности могут быть просмотрены. По локализации кожные изменения не всегда совпадают с костными поражениями. Гиперпигментированные участки кожи не приподняты.
При синдроме Олбрайта встречаются и другие отклонения - гипертиреоз, диабет, пороки сосудов, хрящевая остеодисплазия и др. Биохимические показатели при синдроме Олбрайта нормальны; иногда повышена активность щелочной фосфатазы (до 20 ед. Боданского). Среди осложнений фиброзной дисплазии, специфических для синдрома Олбрайта, встречаются атрофия зрительного нерва и экзофтальм как следствие гиперостоза области турецкого седла и орбиты.
Прогноз при синдроме Олбрайта благоприятный, однако возможны патологические переломы и деформация костей и в редких случаях озлокачествления фиброзных фокусов.
Дифференциальная диагностика и лечение костного компонента синдрома Олбрайта - см. Остеодисплазия. Кожные и эндокринные проявления при синдроме Олбрайта необратимы.
Уплотнение костной структуры основания черепа, преимущественно в области турецкого седла.
Псевдогипопаратиреоз (синдром Олбрайта) - это заболевание, характеризующееся морфологическими изменениями, гипокальциемической тетанией и отсутствием реакции на введение паратгормона. При заболевании отмечается низкорослость, коренастое телосложение, круглое лицо, преждевременное закрытие эпифизарных ядер окостенения, ожирение, брахидактилия, иногда короткие плюсневые кости, дистрофические изменения зубов и ногтей. Иногда наблюдается отставание интеллекта, кальцификация мягких тканей. В отличие от истинного гипопаратиреоза гипокальциемия, гиперфосфатемия и гипофосфатурия не корригируются введением паратгормона.
Заболевают обычно люди молодого и зрелого возраста.
Олбрайт объяснял это состояние рефрактерностью эпителия канальцев почек к действию эндогенного и экзогенного паратгормона, что приводит к повышенной обратной реабсорбции фосфора почками с гиперфосфатемией, гипофосфатурией, гиперкальциемией и гиперкальцийурией. Он относил псевдогипопаратиреоз к другим аналогичным состояниям органной рефрактерности к определенным гормонам (см. Сибрайт-бантамский синдром).
При этом заболевании околощитовидная железа имеет нормальное строение.
Псевдопсевдогипопаратиреоз - описанное в 1952 г. Олбрайтом патологическое состояние, напоминающее псевдогипопаратиреоз с присущими ему морфологическими изменениями, но без соответствующих гуморальных изменений.
Нарушения в эндокринной системе
Чаще всего у девочек, имеющих синдром Олбрайта наблюдается преждевременное половое созревание, которое вызвано эстрогенами, выделяющимися в кровь от кисты яичников. Кисты могут увеличиваться, после уменьшаться в своих размерах в течение нескольких недель или дней. С помощью процедуры УЗИ есть возможность увидеть и измерить размеры новообразований. Кисты могут вырастать до вполне приличных размеров. Были случаи, когда она выросла до размеров мяча для гольфа, то есть больше 50 мм в диаметре.
Увеличение груди и менструальные кровотечения наблюдаются вместе с ростом кисты. Если у девочки началась менструация до 2 лет, то это является первым симптомом синдрома Олбрайта. Однако наличие нерегулярных менструаций и кист яичников могут наблюдаться как у подростков, так и у взрослых женщин. Это все не мешает иметь здоровых детей.
Лечение детей, имеющих преждевременное половое созревание, достаточно трудно и малоэффективно. Даже если кисту удалят хирургическим способом, она может снова возникнуть. При принятии гормона прогестерона может быть остановлена менструация, но быстрые темпы развития и роста костей не замедляются. Возможны негативные последствия для работы надпочечников. При лечении используются препараты для приема внутрь, блокирующие синтез эстрогена.
Функции щитовидной железы
50 процентов людей, имеющих синдром Олбрайта, страдают нарушениями функции щитовидной железы. Это так называемый зоб, узелки и кисты. Возможны в редких случаях тонкие структурные изменения. У этих пациентов уровень вырабатываемого гипофизом тиреотропного гормона низкий, а вот уровни гормонов щитовидной железы имеют нормальные показатели или немного повышены. Проводится лечение, с помощью которого уменьшается синтез гормонов щитовидной железы. Оно показано в тех случаях, когда уровень выделяемых гормонов достаточно высок.

Чрезмерная секреция гормона роста
При заболевании гипофиз начинает выделять большое количество гормона роста. У детей, которым был поставлен такой диагноз, как синдром Олбрайта, была обнаружена акромегалия. У юношей стали появляться грубые черты лица, быстро увеличились руки и ноги, они могли страдать артритом. Лечение детей с такими признаками сводится к хирургическому удалению области гипофиза и использованию синтезированных аналогов гормона соматостатина, подавляющих выработку гормона роста.
Прочие эндокринные нарушения
Достаточно редко имеется чрезмерная секреция и расширение надпочечников. Подобное нарушение может привести к ожирению туловища и лица, увеличению массы тела, прекращению роста и кожной хрупкости. Все эти симптомы назвали синдромом Кушинга. При таких изменениях удаляют пораженный надпочечник или принимают лекарства, которые уменьшают синтез кортизола.
Иногда у детей, которые имеют синдром Олбрайта, в крови наблюдается очень низкий уровень фосфора вследствие большой потери фосфатов с мочой. Это нарушение может быть причиной изменения костей, связанного с рахитом. В качестве лечения назначают пероральные фосфаты и витамин D в дополнение.
Нарушения, связанные с кожей
На коже с рождения или вскоре после него появляются пятна цвета «кофе с молоком». Они чаще всего возникают на крестце, туловище, конечностях, ягодицах, задней поверхности шеи, лбу, на волосистой части головы, затылке. Все они также являются признаком того, что у ребенка синдром Олбрайта. Фото этих пятен можно увидеть ниже.

Хотя при таком заболевании, как нейрофиброматоз, тоже есть пятна цвета «кофе с молоком». Однако для синдрома Олбрайта характерны более крупные пятна с неправильными очертаниями, их меньше по количеству. Они имеют диаметр от 1 до нескольких сантиметров, коричневый оттенок. Цвет у всех одинаковый, они овальные по своей форме, для них характерна гладкая поверхность. При гистологических исследованиях чаще всего выявляется, что эпидермис не изменен по своей структуре, но количество меланина в кератиноцитах немного увеличено.
Единичные пятна подобного типа могут встречаться и у вполне здоровых людей. Если они не беспокоят и не растут, то в лечении нет необходимости. Если же наблюдается интенсивный рост, имеются пятна неправильной формы, то рекомендуется исследовать их гистологически. А затем удалить хирургическим способом.

Заключение
Таким образом, можно сказать, что для синдрома Олбрайта характерно поражение костей или черепа, наличие на коже пигментных пятен, раннее половое созревание. Хотя бывают случаи, когда присутствуют только первые два симптома. Вообще основным признаком синдрома являются костные поражения (остеодисплазия). Однако при наступлении половой зрелости этот процесс приостанавливается. У взрослых людей изменения костей не прогрессируют. Вообще, при выявлении и правильном лечении прогноз терапии данного заболевания вполне благоприятный.
Catad_tema Педиатрия - статьи
Псевдогипопаратиреоз (наследственная остеодистрофия Олбрайта): сложности дифференциально-диагностического поиска. Клиническое наблюдение
Е.В. Тозлиян, педиатр-эндокринолог, генетик, к. м. н., И.В. Шулякова, невролог, к. м. н.,
обособленное структурное подразделение «Научно-исследовательский клинический институт педиатрии» ГБОУ ВПО «Российский национальный исследовательский медицинский университет имени Н.И. Пирогова» Минздрава РФ, г. Москва
Ключевые слова:
дети, псевдогипопаратиреоз, наследственная остеодистрофия Олбрайта, ожирение, гипокальциемия, диагностика, резистентность к паратиреоидному гормону.
Keywords:
children, pseudohypoparathyroidism, Albright hereditary osteodystrophy, obesity, hypocalcemia, diagnostics, parathyroid hormone resistance.
Псевдогипопаратиреоз (греч. pseudes – ложный + гипопаратиреоз; синоним: наследственная остеодистрофия Олбрайта, синдром «яванской курицы») – редкое наследственное заболевание костной системы, имитирующее гипопаратиреоз и характеризующееся нарушением обмена кальция и фосфора; часто сопровождается задержкой умственного и физического развития. Заболевание описано впервые американским врачом-эндокринологом Albright F. в 1942 году . Распространенность заболевания составляет 7,9 на 1 млн человек .
ГЕНЕТИЧЕСКИЕ ДАННЫЕ
Псевдогипопаратиреоз (ПГП) – генетически гетерогенное заболевание. Данные о типе наследственной передачи противоречивы: как X-сцепленный доминантный , так и аутосомно-доминантный, аутосомно-рецессивный типы . В большинстве случаев развитие наследственной остеодистрофии Олбрайта связано с мутациями в расположенном на хромосоме 20 локусе 20q13 гена GNAS1 (Patten et al., 1990), кодирующего белок Gs-альфа, связанного с рецептором паратиреоидного гормона (ПТГ) . Подобный фенотип выявлен и у больных с интерстициальной делецией длинного плеча хромосомы 2 локуса 2q37 .
ПАТОГЕНЕЗ
В основе патогенеза псевдогипопаратиреоза лежит генетически обусловленная резистентность почек и скелета к действию парат-гормона в результате дефекта комплекса «специфический циторецептор – паратгормон – аденилатциклаза», что нарушает процесс образования в почках циклического 3"-, 5"-аденозинмонофосфата (цАМФ), являющегося внутриклеточным посредником действия паратгормона на метаболические процессы. Псевдогипопаратиреоз является генетически гетерогенным заболеванием. У части больных дефектен сам циторецептор, связывающий паратгормон (тип 1А псевдогипопаратиреоза), у других отмечается дефект нуклеотидсвязывающего белка, локализованного в липидном бислое клеточной мембраны и функционально связывающего рецептор с аденилатциклазой (тип 1B псевдогипопаратиреоза). У некоторых больных наблюдается ферментативная недостаточность самой аденилатциклазы (псевдогипопаратиреоз 2-го типа). Дефицит цАМФ, получающийся вследствие этих дефектов, ведет к нарушению синтеза специфических белков, определяющих биологический эффект паратгормона. Таким образом, теряется чувствительность органов-мишеней к паратгормону .
КЛИНИЧЕСКАЯ ХАРАКТЕРИСТИКА
В настоящее время выделяют 4 клинические формы патологии: типы 1А, 1В, 1С и 2. Знание их клинико-биохимических особенностей и данных генетических исследований позволяет провести дифференциальную диагностику в рамках самой нозологической формы.
Общими признаками, позволяющими заподозрить заболевание, являются диспропорциональность физического развития, низкий рост (до карликовости) за счет укорочения нижних конечностей (фото 1), брахидактилия (фото 2), круглое «лунообразное» лицо (фото 3). Иногда наблюдаются экзостозы и аплазия зубов.
Фото 1.
Внешний вид ребенка с остеодистрофией Олбрайта
(особенности фенотипа, низкий рост за счет укорочения нижних конечностей)

Фото 2.
Особенности костной системы у больного
с остеодистрофией Олбрайта
(брахидактилия – укорочение пальцев)
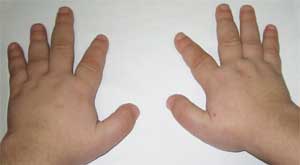
Фото 3.
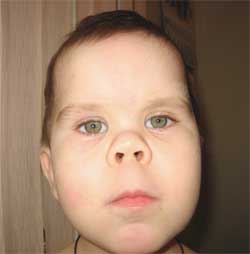
Особенности фенотипа ребенка
с остеодистрофией Олбрайта
(круглое «лунообразное» лицо)
Патогномоничным признаком считается резкое укорочение I, III и V пястных и плюсневых костей (особенно III и IV), вследствие чего II пальцы на кистях и стопах оказываются длиннее остальных, а при сжатии кисти в кулак отсутствуют выпуклости в области IV и V пястно-фаланговых суставов – так называемый брахиметафалангизм. Выявляются также короткие широкие фаланги, утолщение свода черепа и деминерализация костей (остеопороз), ожирение .
Умственная отсталость (чаще умеренной степени выраженности) обнаруживается примерно у 20% больных. По данным некоторых авторов , олигофрения встречается в 70% случаев при наличии гипокальциемии и в 30% случае при нормокальциемии. Психические процессы у больных замедлены. В неврологическом статусе нередко отмечаются моторная неловкость, невротические реакции: страхи, тревога, беспокойство, плохой сон, повышение рефлексов, судороги, носящие тетанический характер и обусловленные гипокальциемией, иногда судорожные пароксизмы. Описаны также миопатические симптомы: мышечная утомляемость, мышечная слабость. Часто наблюдаются экстрапирамидные нарушения: хореиформные гиперкинезы, атетоз, лицевой гемиспазм, паркинсонизм, в отдельных случаях имеют место эпилептические пароксизмы, мозжечковые симптомы: атаксия, нарушение координации.
Нередко определяются кальцификация мягких тканей, подкожные кальцификаты (грудь, живот, пяточные сухожилия), при гистологическом исследовании которых – osteoma cutis (Izraeli et al., 1992), мозге (базальные ганглии). Важно отметить, что кальцификаты могут быть уже при рождении. Вследствие гипокальциемии обычно развивается катаракта и возникают дефекты эмали зубов.
ПСЕВДОГИПОПАРАТИРЕОЗ ТИПА 1А
имеет аутосомно-доминантный тип наследования. Ген псевдогипопаратиреоза типа 1А – GNAS1 – локализован на длинном плече хромосомы 20, в локусе 20q13.2. Развитие заболевания связано с дефицитом гуанин-нуклеотид-связывающего белка (Gs-белок). При этом ПТГ, связываясь с рецепторами тканей-мишеней, не способен активизировать циклический аденозинмонофосфат (цАМФ) и вызвать тканевой ответ. Вероятно, подобный механизм лежит в основе развития нечувствительности тканей других органов и эндокринных желез (гипофункция щитовидной железы, гонад, гипофиза, сахарный диабет, а также сниженный ответ печени на введение глюкагона), наблюдаемой при псевдогипопаратиреозе типа 1А. При данном типе патологии не наблюдается характерной для нормы повышенной экскреции цАМФ с мочой в ответ на экзогенное введение ПТГ. Заболевание диагностируется чаще в возрасте 5–10 лет. У больных наблюдаются низкий рост, короткая шея, круглое лицо, укорочение метакарпальных и метатарзальных костей (чаще укорочение IV и реже II пальцев) – так называемый брахи-метафалангизм. Отмечаются кальцификация мягких тканей, подкожные кальцификаты, которые могут выявляться уже при рождении; нередко наблюдается одновременное вовлечение других эндокринных желез: щитовидной железы (гипофункция), гонад, поджелудочной железы (сахарный диабет). Вследствие гипокальциемии нередко развиваются катаракта и дефект эмали зубов. В качестве дифференциально-диагностического теста отличия ПГП типа 1А от гипопаратиреоза: отсутствие клинического эффекта от парентерального введения ПТГ в виде подъема уровня кальция в крови и увеличения почечной экскреции фосфора с мочой (фосфатурический эффект).
При биохимическом исследовании выявляются гипокальциемия, гиперфосфатемия, увеличение уровня паратиреоидного гормона в крови, гипофосфатурия. Уровень Gs-белка в крови снижен. При рентгенологическом исследовании костной системы обнаруживаются укорочение метакарпальных и метатарзальных костей, генерализованная деминерализация, утолщение костей свода черепа.
ПСЕВДОГИПОПАРАТИРЕОЗ ТИПА 1B
имеет аутосомно-доминантный тип наследования, однако не исключен доминантный, сцепленный с Х-хромосомой тип наследования. Необходимо иметь в виду наблюдающуюся иногда неполную пенетрантность гена болезни и возможность скрытого носительства патологии. Поэтому рекомендуется клиническое (выявление субклинического течения болезни) и биохимическое обследование (определение уровней кальция, фосфора, ПТГ крови) предполагаемых носителей заболевания. ПГП типа 1В обусловлен дефицитом тканевых рецепторов к паратиреоидному гормону в органах-мишенях и ограниченной резистентностью к паратгормону. Клиническая картина сходна с клиникой типа 1А, но отсутствует поражение других эндокринных желез, реже встречается остеодистрофия.
У больных отсутствует реакция почек на экзогенное введение паратиреоидного гормона в виде увеличения экскреции циклического аденозинмонофосфата с мочой, однако, в отличие от типа 1А, уровень Gs-белка в крови нормален. Женщины поражаются чаще мужчин, однако тяжесть заболевания может быть одинаковой как у мужчин, так и у женщин.
ПСЕВДОГИПОПАРАТИРЕОЗ ТИПА 1С
некоторые авторы отождествляют с псевдо-псевдогипопаратиреозом (ППГП), описанным Albright F. в 1952 году. Характеризуется свойственной ПГП клинической картиной, однако уровни кальция, фосфора в крови и моче остаются в пределах нормы. Показатели ПТГ и Gs-белка в крови также сохраняются на нормальном уровне. У некоторых больных с ПГП типа 1С обнаруживаются делеции de novo на хромосоме 2 . Не исключено, что этот вариант болезни является подтипом ПГП типа 1А.
ПСЕВДОГИПОПАРАТИРЕОЗ ТИПА 2
клинически сходен с другими типами заболевания, однако имеет аутосомно-рецессивный тип наследования. Не исключено существование и аутосомно-доминантных форм патологии. Патогенез развития связан с внутриклеточной резистентностью к цАМФ. ПТГ при этом связывается с рецепторами и вызывает нормальную ответную реакцию клеток на ПТГ в виде увеличения экскреции цАМФ. Внутриклеточная нечувствительность к цАМФ, однако, не позволяет осуществиться полной реализации действия ПТГ. При этом сохраняется нормальная реакция почек на экзогенное введение паратиреоидного гормона в виде увеличения экскреции циклического аденозинмонофосфата с мочой. Высказывается мнение, что ПГП типа 2 может быть связан с дефицитом витамина D .
Таким образом, выделенные типы ПГП клинически характеризуются пониженной чувствительностью органов-мишеней к ПТГ, однако различаются патогенетическими механизмами формирования нечувствительности тканей.
ДИАГНОСТИКА
Лабораторным дифференциально-диагностическим тестом может служить характер почечной экскреции цАМФ в ответ на введение ПТГ: повышенная экскреция цАМФ отмечается при типе 2 и ее отсутствие – при типе 1. Диагноз подтверждается обнаружением сниженного уровня гуанин-нуклеотидсвязывающего белка (Gs-белок) в крови (в среднем в 1,5–2 раза) по сравнению с нормой. Гипокальциемия, как правило, сочетается с гиперфосфатемией и гипофосфатурией. Уровень ПТГ повышен; при 1C-типе уровень ПТГ в норме, что дало основание для названия «псевдогипопаратиреоз». При рентгенологическом исследовании костной системы обнаруживаются укорочение пястных и плюсневых костей, нередко генерализованная деминерализация (остеопороз), утолщение костей свода черепа. В дерматоглифическом рисунке отмечается смещение осевого ладонного трирадиуса.
Критерии диагноза:
- низкий рост;
- круглое лицо;
- задержка нервно-психического развития;
- скелетные аномалии;
- низкое содержание кальция в сыворотке крови;
- высокий уровень паратиреоидного гормона в крови;
- снижение экскреции с мочой фосфатов и цАМФ.
ЛЕЧЕНИЕ И ПРОФИЛАКТИКА
Лечение при гипокальциемии заключается в назначении препаратов кальция в дозах, достаточных для поддержания нормальной концентрации кальция в крови. Большое значение имеет терапия витамином D. В настоящее время применяют активные метаболиты витамина D – оксидевит, 1-альфа-Д3, кальцитрин и др. в дозе 1–2 мкг/сутки с положительным результатом (увеличение содержание кальция в крови, уменьшение проявлений судорожного синдрома). Эффективен также тахистин (0,5–1,5 мг/сутки). Данный препарат увеличивает всасывание кальция в кишечнике и тем самым способствует повышению уровня кальция в крови. Противосудорожная терапия используется как дополнительное лечение. На интеллектуальное развитие лечение не оказывает заметного действия, но наряду с уменьшением симптомов судорожного синдрома наблюдается регресс неврологических проявлений (подкорковых нарушений, хореиформных гиперкинезов, атетоза и др.). Во избежание передозировки препаратов витамина D необходим контроль концентрации кальция в крови каждые 3–7 дней в течение первых 2 недель лечения и каждый месяц в течение последующих 2–3 месяцев. По достижении стабильной концентрации кальция в крови достаточно проверять ее 1 раз в 2–3 месяца. Диета с ограничением фосфора помогает нормализовать как концентрацию фосфора, так и содержание кальция в крови и устранить симптомы вторичного гиперпаратиреоза. При недостаточности других желез внутренней секреции проводят заместительную терапию соответствующими гормонами.
Лечение паратгормоном неэффективно. Для купирования судорожных приступов внутривенно вводят 10%-ный раствор кальция хлорида или кальция глюконата; внутрь – 5–10%-ный раствор кальция хлорида по 1 столовой ложке 3–4 раза в день: кальция глюконат, кальция лактат – до 10 г в день.
ПРОГНОЗ для жизни определяется выраженностью судорожного синдрома.
ПРОФИЛАКТИКА болезни основывается на данных медико-генетического консультирования.
МЕДИКО-ГЕНЕТИЧЕСКОЕ КОНСУЛЬТИРОВАНИЕ
При медико-генетическом консультировании следует исходить из аутосомно-доминантного типа наследования и высокого (50%) риска повторения заболевания в семье при унаследованных формах. С целью идентификации характера типа наследования необходимо проводить тщательное обследование родителей, так как синдром может проявляться минимальными клиническими симптомами. В настоящее время разработана и совершенствуется молекулярно-генетическая диагностика заболевания путем типирования мутаций в гене GNAS1 на хромосоме 20. Разрабатываются способы пренатальной диагностики заболевания в целом и отдельных его типов.
КЛИНИЧЕСКОЕ НАБЛЮДЕНИЕ Мальчик Г., 14,5 лет (фото 4), поступил в Научно-исследовательский клинический институт педиатрии с диагнозом: дегенеративное заболевание нервной системы? врожденная наружная гидроцефалия; симптоматическая эпилепсия; наследственный синдром? болезнь накопления? метаболическая энцефалопатия; субклинический гипотиреоз; низкорослость смешанного генеза; когнитивные нарушения.
Жалобы при поступлении на интенсивные приступообразные головные боли, локализующиеся в лобной области и сопровождающиеся рвотой, которая приносит облегчение, снижение памяти и успеваемости в школе, судорожные приступы, во время которых происходит подергивания в правой руке.
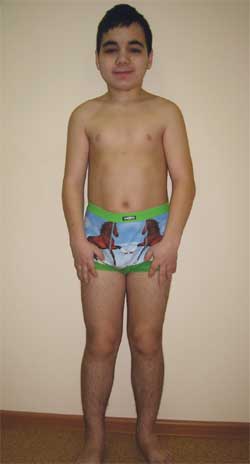
Фото 4.
Ребенок Г., 14,5 лет, с остеодистрофией Олбрайта
(особенности фенотипа, низкий рост, укорочение конечностей, брахидактилия)
Анамнез семейный: родители – армяне по национальности, не состоящие в кровном родстве и не имеющие профессиональных вредностей. В родословной случаев психических заболеваний, эпилепсии, задержки в развитии – не отмечалось. Сибс, сестра 17 лет – со слов – здорова.
Анамнез жизни и заболевания:
мальчик от 2-й беременности, протекавшей без особенностей, роды вторые, в срок, физиологические, масса при рождении – 3100 г, длина – 51 см. Закричал сразу, оценка по шкале Апгар – 7/9 баллов. Ухудшение состояния на 3-и сутки – судороги неонатальные, купированы в роддоме. Ранний постнатальный период – без особенностей. Отмечалась незначительная темповая задержка моторного развития на первом году жизни, самостоятельная ходьба с 1 года 3 мес. В связи с чем наблюдался неврологом с диагнозом: органическое поражение ЦНС; врожденная гидроцефалия; неонатальные судороги; фебрильные судороги в анамнезе.
Получал диакарб, финлепсин. Дебют приступов с 1 года 11 мес. – асимметричные, тонические в виде напряжения правой руки и ноги, с заведением глаз, до 2 мин., без потери сознания, частые до 10 эпизодов в сутки. Получал депакин нерегулярно. На фоне самостоятельной отмены – однократный тонический статус. В 2 года проведена по месту жительства КТ головного мозга, где выявлены единичные очаги демиелинизации в затылочных долях.
Консультирован нейрохирургом, рекомендовано консервативное лечение. С 3 лет отмечается задержка психоречевого развития, рекомендовано наблюдение психиатра.
С 4–5 лет родители стали отмечать деформацию и укорочение пальцев стоп и кистей, в особенности II–IV пальцев симметрично на руках и ногах, снижение ростовых показателей. В 8 лет заключение логопеда – общее нарушение речи 2–3-го уровня, рекомендовано обучение в специализированной школе. В этом же возрасте осмотр генетиком по месту жительства, заключение: наследственная болезнь обмена? рекомендовано исследование аминокислот крови, изменений не было выявлено; окончательное заключение: данных за наследственное заболевание обмена не выявлено; гипохондроплазия; рекомендовано лечение невролога и эндокринолога.
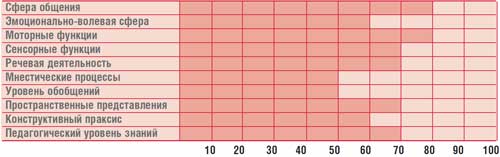
Таблица.
Профиль психического развития ребенка Г., 14,5 лет (IQ = 68)
В возрасте 8 лет консультирован эндокринологом по поводу задержки роста и развития. При рентгенологическом исследовании кистей рук отмечены особенности: средние, основные фаланги и пястные кости укорочены, утолщены; диагноз рентгенолога – ахондроплазия.
Неоднократно обследуется по месту жительства в неврологическом стационаре. В 12 лет появились судорожные приступы без потери сознания с подергиванием правой руки, носящие серийный характер, назначена противосудорожная терапия (депакин), частота приступов значительно сократилась. В 13 лет проведена МРТ головного мозга с контрастированием – симметричные изменения в основании височных долей на уровне ядер в виде повышения МР-сигнала, что характерно для токсических (марганец) или метаболических (медь, железо) энцефалопатий.
Вновь осмотрен в возрасте 13 лет 3 мес. эндокринологом, при исследовании тиреоидного профиля выявлено повышение тиреотропного гормона (ТТГ), диагностирован субклинический гипотиреоз, назначен L-тироксин.
При анализе амбулаторной карты ребенка и документации по месту жительства исследование кальция и фосфора проводилось однократно, в возрасте 1,5 лет, отмечалась гипокальциемия, но по данному поводу дообследование не проводилось. Учитывая неопределенность диагноза по месту жительства, генетиком ребенок направлен в Москву, в Научно-исследовательский клинический институт педиатрии, с целью уточнения диагноза.
Данные объективного исследования:
Рост – 143 см, масса – 43 кг.
Физическое развитие очень низкое, гармоничное, телосложение диспропорциональное за счет укорочения конечностей. Sds роста соответствует –2,8 отклонениям от нормы (норма –2+2).
Особенности фенотипа: круглое лицо, короткая шея, антимонголоидный разрез глазных щелей, широкое переносье, высокий лоб, брахидактилия, укорочение IV и V пястных и плюсневых костей (фото 5). По внутренним органам – без особенностей. Половое развитие – Tanner III–IV стадия (что соответствует возрасту).
Данные лабораторных и функциональных исследований:
Клинический анализ крови и мочи – норма.
Биохимический анализ крови: общий кальций – 1,39 (норма 2,02–2,6 ммоль/л), кальций ионизированный – 0,61 (норма 1,13–1,32 ммоль/л), фосфор неорганический – 3,66 (норма 0,86–1,56 ммоль/л), остальные показатели в пределах нормы.
Биохимический анализ мочи: почечная экскреция фосфатов снижена – 11,5 ммоль/л (норма 19–32 ммоль/л).
Тиреоидный профиль: ТТГ – 11,75 (норма 0,4–4,0 мкМЕ/мл), свободный Т4 – 0,49 (норма 1,0–1,8 нг/дл).
Паратиреоидный гормон – 499 (норма 12– 65 пг/мл), СТГ – 7 нг/мл (норма 7–10 нг/мл), соматомедин-С – 250 нг/мл (норма 88–360 нг/мл).
УЗИ внутренних органов – без особенностей.
ЭКГ – миграция суправентрикулярного водителя ритма на фоне регулярной ЧСС 71– 80 уд/мин. Неполная блокада правой ножки пучка Гиса. Нарушение процесса реполяризации в миокарде задней стенки левого желудочка (снижение з.Т III, аVF).
R-графия позвоночника – правосторонний сколиоз грудного отдела позвоночника 1-й степени, выраженный остеопороз.
R-графия кистей рук с захватом предплечий – укорочение и расширение концевых и средних фаланг. Костный возраст – 13,5– 14 лет.
ЭЭГ-паттернов эпилептической активности не зарегистрировано.
МРТ головного мозга – МР-картина множественных субкортикальных очагов повышенного МР-сигнала в лобных долях, наружная компенсированная гидроцефалия с атрофией вещества головного мозга.
МСКТ головного мозга – симметричные участки обызвествления лентиформных ядер. Диффузные гиперденсивные участки в таламусах, хвостатых ядрах с участком обызвествления справа. Множественные точечные обызвествления покровных мягких тканей черепа.
Аудиограмма – без патологии.
ДНК-диагностика в гене GNAS1 – в работе.
Консультации специалистов:
Эндокринолог – наследственная остеодис-трофия Олбрайта типа 1А (псевдогипопара-тиреоз). Первичный гипотиреоз, неполная медикаментозная компенсация.
Окулист – катаракта полная вторичная. Рекомендовано оперативное лечение.
Психолог – когнитивные нарушения (психологический профиль ребенка представлен в табл.).
Учитывая фенотип ребенка, данные анамнеза, результаты дополнительных исследований (гипокальциемия, гиперфосфатемия, гипофосфатурия, повышение паратиреоидного гормона крови), кальцинаты в веществе головного мозга, наличие катаракты, гипотиреоза), поставлен диагноз: наследственная остеодистрофия Олбрайта 1А-типа (псевдогипопаратиреоз). Рекомендовано проведение ДНК-диагностики - поиск мутаций в гене GNAS1.
Лечение: ребенку рекомендован прием эутирокса в дозе 100 мкг/сутки; активный метаболит витамина Д - альфа-Д3 («Тева») в дозе 2 мкг/сутки; кальций («Сандоз») 2000 мг/сутки; постоянный прием противосудорожной терапии - финлепсин 800 мг/сутки под наблюдением невролога-эпилептолога; занятия с логопедом-дефектологом и психологом; энерготропная терапия (Элькар и коэнзим Q10 в возрастных дозах). Контроль показателей фосфорно-кальциевого обмена, уровня паратгормона.
Таким образом,
представленное клиническое наблюдение демонстрирует сложности дифференциально-диагностического поиска, важность своевременного исследования простых биохимических параметров (при эпилепсии обязателен неоднократный скрининг показателей фосфорно-кальциевого обмена), исходы поздней диагностики генетически детерминированного заболевания, необходимость интегрировать отдельные признаки в общий фенотип того или иного патологического состояния для целенаправленной своевременной диагностики отдельных форм наследственных заболеваний. Своевременная диагностика, уточнение генеза каждого синдрома особенно важны, так как позволяют найти оптимальный подход к лечению этих состояний, профилактике возможных осложнений (вплоть до инвалидности ребенка); предупреждение повторного возникновения наследственных болезней в пораженных семьях (медико-генетическое консультирование). Это диктует необходимость врачам различных специальностей четко ориентироваться в потоке наследственно обусловленной патологии. Список литературы находится в редакции.
Статьи по теме















