血液凝固スキームの段階 生化学
いいね。 紡績システム
血
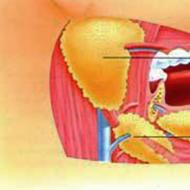
血管が損傷を受けると、一連の反応が開始され、その結果、血栓が形成されます。これは、出血を防ぐ血栓です。 血液凝固(凝固)における主な役割は、血小板およびいくつかの血漿タンパク質によって果たされています。
出血を止めるには3つの段階があります。 最初の段階で、血管は縮小しています。 その後、血小板が損傷部位に付着し、それらが互いに重なり合って血小板栓(白い血栓)を形成する。 白い血栓は壊れやすく、小さな血管を塞ぐことしかできません。 第三段階では、血漿の可溶性タンパク質であるフィブリノゲンが、血小板間に沈着する不溶性フィブリンタンパク質に変換され、そして強力なフィブリン血栓が形成される。 このような血栓は赤血球を含んでいるため、赤血球と呼ばれます。
フィブリン血栓の形成は、酵素トロンビンの活性化を導くカスケードのタンパク質分解反応によって先行され、それはフィブリノーゲンをフィブリンに変える。 血液凝固に関与するすべてのタンパク質は凝固因子と呼ばれています。 それらは主に肝臓および血球中でローマ数字で示される不活性前駆体の形態で合成されるが、それらはまた些細な名前を有する(表14−1)。 これらのタンパク質のほとんどは、一連の酵素的血液凝固反応において活性化されています。 アクティブ
表14-1 凝固因子の主な機能と血漿中濃度
| 要因 | T rivial name | 血漿中の含有量、g / l | 機能 |
| 1 | 2 | 3 | 4 |
| 私は | フィブリノーゲン | 2-4 | 可溶性フィブリン前駆体タンパク質 |
| イア | フィブリン | フィブリンゲルを形成する | |
| II | プロトロンビン | 0,1 | プロ酵素* |
| IIa | トロンビン | フィブリノーゲンをフィブリンに変換するプロテアーゼおよび活性化因子V、VII、VIII、XIII、C | |
| III | 組織因子 | タンパク質活性化剤膜複合体VIIa-TF-Ca 2+ | |
| IV | Ca 2+ | 0.9〜1.2 mmol / l | 凝固促進経路の酵素とホスファチジルセリンとの相互作用を仲介する |
| V | プロアセリン | 0,01 | タンパク質 - アクチベーター膜複合体Xa-Va-Ca 2+の前身 |
| Va | アクセリン | 膜複合体Xa-Va-Ca 2+のタンパク質活性化剤 | |
| VII | プロコンベルチン | 0,005 | プロ酵素* |
| VIIa | コンバチン | プロテアーゼ*、活性化因子XおよびIX | |
| Viii | 不活性な抗血友病因子A(不活性な抗血友病グロブリン) | 0,01-0,02 | タンパク質 - アクチベーター膜複合体IXa-VIIIa-Ca 2+の前身 |
| VIIIa | 活性型抗血友病因子A(活性型抗血友病グロブリン) | 膜複合体IXa-VIIIa-Ca 2+のタンパク質活性化剤 | |
| 1倍 | 不活性抗血友病因子B(不活性クリスマス因子) | 0,003 | プロ酵素* |
| イクサ | 活性型抗血友病因子B(活性型クリスマス因子) | X因子を活性化するプロテアーゼ* | |
| X | 無効要因スチュアートプルア | 0,01 | プロ酵素* |
| Xa | Stuart Prouerのアクティブファクター | プロテアーゼ*、活性化因子II | |
| 西 | 不活性血漿トロンボプラスチン前駆体 | 0,005 |
表14-1の続き
| 1 | 2 | 3 | 4 |
| 夏 | 活性血漿トロンボプラスチン前駆体 | プロテアーゼ活性化因子IX | |
| Xii | 不活性なHagemanファクター | 0,03 | 補酵素接触経路酵素 |
| Xii | アクティブファクターHageman | プロテアーゼ、活性化因子XI、プレカリクレイン、プラスミノーゲン | |
| XIII | 不活性トランスグルタミダーゼ(不活性フィブリン基礎化因子) | 0,01-0,02 | プロ酵素 |
| Hsh | 活性型トランスグルタミダーゼ(活性型フィブリン安定化因子) | フィブリン - モノマー、フィブリンおよびフィブロネクチンの分子間のアミド結合の形成を触媒する | |
| プレカシュクレイン | 0,05 | 補酵素接触経路酵素 | |
| カリクレイン | プロテアーゼ、活性化因子XII、プラスミノーゲン | ||
| VMK | 0,06 | 血液凝固接触経路のタンパク質活性化剤 |
*凝固促進剤凝固経路の膜酵素複合体の形成に必要なカルボキシグルタミン酸残基を含む。
これらのタンパク質の形態は、同じローマ数字によって示されるが、文字「a」が追加されている。
A.フィブリン血栓の形成
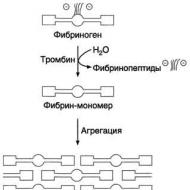
フィブリン血栓の形成は、血漿フィブリノーゲンの可溶性タンパク質の不溶性フィブリンへの変換から始まる。
フィブリノーゲン(第I因子)は分子量340kDの糖タンパク質である。 それは肝臓で合成され、8.02 - 12.9 µmol / L(2 - 4 g / L)の濃度で血漿に含まれています。 フィブリノーゲン分子は、ジスルフィド結合によって互いに結合している6本のポリペプチド鎖からなる。 フィブリノーゲン分子のポリペプチド鎖の組成は、α2、β2、γ2として示され、大文字は、フィブリノーゲンのフィブリンへの変換中にトロンビンの作用下で切断される領域に対応する。 鎖Aβ中のフラグメントAおよび鎖Bβ中のBは、大量のアスパラギン酸およびグルタミン酸残基を含有する。 これにより、フィブリノーゲン分子のN末端に強い負電荷が生じ、それらの凝集が妨げられる。
フィブリノーゲン分子は3つの球状ドメイン、分子の各末端に1つ(ドメインD)および中央に1つ(ドメインE)からなる。 ドメインは、棒状配置を有するポリペプチド鎖の部分によって互いに分離されている。 鎖AαおよびBβのN末端フラグメントAおよびBは中央ドメインEから突き出ている(図14−8)。
フィブリン血栓の形成において、4つの段階が区別され得る。
フィブリノーゲンのフィブリンモノマーへの変換。 第一に、フィブリノーゲン分子が負に帯電したフラグメントAおよびBから放出され、その結果としてフィブリンモノマーが形成される。 フィブリノーゲン(第I因子)からフィブリン(第1a因子)への変換は、酵素トロンビン(第Pa因子)によって触媒される。 フィブリノーゲンの各分子において、トロンビンは4つのペプチド結合アルギニルグリシルを加水分解し、そのうち2つはフラグメントAをα鎖に結合する。
図 14−8。 フィブリノーゲンの構造 フィブリノーゲンは、6つのポリペプチド鎖、すなわち、α2、β2およびγ2からなる。 A、B - フィブリノーゲン分子が凝集しないために負に帯電したフラグメント。 D、E - フィブリノーゲン分子の球状ドメイン。 ドメインは、棒状配置を有するポリペプチド鎖の部分によって分離されている。 中心球状ドメインEから、鎖Aα2およびBβ2のフラグメントAおよびBのN末端部分が突き出ている。
他の2つはフィブリノーゲンのAα2およびββ2鎖にβ鎖を有するBである。 フィブリノーゲンから形成されるフィブリンモノマーは組成(α、β、γ)2を有する。
2. 不溶性フィブリンゲルの形成 第二段階では、不溶性ポリマーフィブリンクロット - フィブリンゲルが形成される。 フィブリノーゲンのフィブリンモノマーへの変換の結果として、ドメインEは結合部位をドメインDに開く。さらに、ドメインEはトロンビンの作用下でのフィブリノーゲンの部分的タンパク質分解後にのみ形成される凝集中心を含み、そしてドメインDは永久凝集中心の担体である。 フィブリン分子の一次凝集は、ある分子のEドメインの結合部位と他の分子のDドメイン上の相補的領域との相互作用の結果として起こり、したがって、非共有結合がフィブリン - モノマー分子のドメイン間に形成される。 フィブリンゲルが「自己集合」すると、最初に二重縫合プロトフィブリルが形成され、ここでフィブリン分子は互いに対して1/2の長さだけ変位している。 プロトフィブリルが特定の臨界長に達すると、それらの側方会合が始まり、太いフィブリン繊維が形成される(図14-9)。 得られたフィブリンゲルは、その中のフィブリン分子が非共有結合によって相互連結されているので、壊れやすい。
3. フィブリンゲル安定化 あるフィブリン分子のリジン残基と他の分子のグルタミン残基との間にアミド結合が形成される結果として、フィブリンゲルは安定化する。 トランスアミダーゼ酵素は、トランスグルタミダーゼ酵素(第XIIIa因子)によって触媒される(図14-10)。 第XIII因子はトロンビンによる部分的タンパク質分解によって活性化される。
トランスグルタミダーゼはまた、フィブリンとフィブロネクチン、細胞外マトリックスの糖タンパク質および血漿との間にアミド結合を形成する(セクション15を参照のこと)。 このようにして、血栓が血管の損傷部位に固定される。
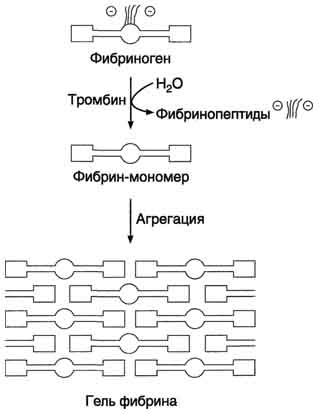
図 14−9。 フィブリンゲル形成 負に帯電したフラグメント(フィブリノペプチド2Aおよび2B)からのトロンビンから放出されたフィブリノーゲンは、フィブリンモノマーに変換される。 フィブリン - モノマーのE-およびD-ドメインの相補的領域の相互作用の結果として、分子の最初の線状および次に横方向の重合がフィブリンゲルの形成と共に起こる。

フィブリンクロットの引き込み。 ゲルの圧縮(収縮)はアクトミオシン血小板、すなわちATPアーゼ活性を有する収縮タンパク質トロンボステニンを提供する。 トロンボステニンは血小板の活性化と凝集にも関与しています。 血栓の収縮は血管の完全な閉塞を防ぎ、血流を回復させる可能性を生み出します。
凝血塊形成のメカニズムには、機能的に異なる3つの段階があります。凝血促進経路、接触経路、および凝血塊の拡大を防ぐ抗凝血相です。
血液凝固のプロコアグラント法
毛細血管および血管からの出血を止めるために、失血を防ぐ血栓の迅速な形成が必要である。 これは、多くの段階で増幅メカニズムを伴うカスケードの酵素反応によって達成される。
凝血促進経路は血液凝固の中心である(図14-11)。
循環血液には、タンパク質分解酵素のプロ酵素が含まれています:第II因子(プロトロンビン)、第VII因子(プロコンベルチン)、第IX因子(クリスマス)、第X因子(Stewart)。 血液中の第Va因子(アクセレリン)および第VIIIa因子(抗血友病因子)ならびに膜タンパク質 - 組織因子(TF、第III因子)はこれらの酵素の活性化タンパク質である(表14−1)。
血管が損傷を受けると、酵素活性化のカスケードメカニズムは、細胞膜リン脂質に関連する3つの酵素複合体の連続的形成によって活性化される。 各複合体は、タンパク質分解酵素、活性化タンパク質およびCa 2+イオンからなる:VIIa-TO-Ca 2+、IXa-VIIIa-Ca 2+(テナーゼ)、Xa-Va-Ca 2+(プロトロンビナーゼ)(図14-)。 12)。 Xa-Va-Ca 2+複合体(プロトロンビナーゼ複合体)はプロトロンビンを活性化する(第II因子)。 一連の酵素反応は、フィブリンモノマーの形成とそれに続く血栓の形成で終わる。
カスケード酵素の活性化においては、3つの主要なメカニズム、すなわち部分的タンパク質分解、活性化タンパク質との相互作用、および修飾細胞膜との相互作用が区別される。
部分的タンパク質分解による活性化。凝固促進性経路のすべての酵素はセリンプロテアーゼであり、不活性な前酵素として肝臓で合成され、そしてこの形態で血中を循環する。 血栓形成シグナルを実行する過程において、プロ酵素(VII、IX、XおよびII因子)は部分的タンパク質分解によって活性酵素に変換される。
トロンビン(Pa因子)- 分子量39 kDの糖タンパク質。 それは不活性プロトロンビン前駆体から血液中に形成される。 プロトロンビンは肝臓で合成され、70 kDaの分子量を持ち、残基を含みます。
図 14−11。 凝固促進剤凝固経路 →血液凝固因子の活性化 ・・・正のフィードバックに基づく凝固因子の活性化 酵素複合体の膜リン脂質成分。 額縁タンパク質活性化剤は枠で囲まれている。 1,2 - 第VIIa因子膜複合体VIIa-TF-CA 2+は第IX因子およびX因子を活性化する。 3−第IXa因子膜複合体IXa − VIIIa − Ca 2+は第X因子を活性化する。 Xa − Va − Ca 2+膜複合体の4,5−第Xa因子はプロトロンビン(第II因子)をトロンビン(第IIa因子)に変換し、そして第VII因子を活性化する。 6−10−トロンビン(第IIa因子)は不溶性フィブリノーゲンを可溶性フィブリンに変換し、第VII、VIII、VおよびXIII因子を活性化する。
γ-カルボキシグルタミン酸。 血中のこのタンパク質の濃度は通常0.1 g / lです。 それはXa-Va-Ca 2+膜酵素複合体上に固定され、一方ではγ-カルボキシグルタメート残基をCa 2+と相互作用し、そして他方では活性化タンパク質Vaと直接相互作用する。 従って、酵素反応が起こるための最良の立体条件が作り出される。 第Xa因子は、プロトロンビン分子中の2つのペプチド結合を加水分解する。 その結果、1本のジスルフィド結合で結合した2本の鎖(軽鎖と重鎖)からなるトロンビン分子が形成されます(図14-12)。 トロンビン分子はγ-カルボキシグルタメート残基を含まず、プロトロンビナーゼ複合体から放出される。 部分タンパク質分解によるトロンビンは、フィブリノーゲンをフィブリンに変換し、そして第VII因子、第VIII因子、第V因子、第XIII因子を活性化する。
トロンビンは多くの重要な生理学的機能を果たす:それは血液凝固の凝固促進剤および接触経路の酵素であり、抗凝固相の反応を開始し、血小板凝集を引き起こしそして細胞増殖および修復に関与する分裂促進作用を有する。
第V因子および第VIII因子はまた、部分タンパク質分解によっても活性化され、それぞれ第Va因子および第VIIIa因子に変わる。 これらの因子の活性化の結果として、それらの立体配座変化およびそれらが活性化する膜リン脂質および酵素に対する親和性が増加する。
蛋白質活性化剤と蛋白質分解酵素との相互作用組織因子、第Va因子および第VIIIa因子は、膜リン脂質ならびにそれぞれ酵素VIIa、IXaおよびXaに対する結合部位を有する。 立体配座の変化の結果として活性化タンパク質に結合すると、これらの酵素の活性は増加する。
組織因子(ファクターIII)タンパク質とホスファチジルセリンからなる複合体です。 組織因子のタンパク質部分(アポタンパク質III)は、多くの細胞(脳、肺、肝臓、脾臓など)の表面に露出しており、ホスファチジルセリン原形質膜と会合している。 しかしながら、細胞表面上のアポタンパク質IIIの出現

図 14−12。 プロトロンビナーゼ複合体の第Xa因子によるプロトロンビンの蛋白質分解的活性化 ![]() - カルボキシグルタミン酸の残基。 矢印はプロトロンビン分子中で加水分解されたペプチド結合の位置を示す。 プロトロンビン分子は単一のポリペプチド鎖からなり、そしてプロトロンビンの部分タンパク質分解によって形成されたトロンビンは単一のジスルフィド結合によって一緒に結合した2つのポリペプチド鎖からなる。
- カルボキシグルタミン酸の残基。 矢印はプロトロンビン分子中で加水分解されたペプチド結合の位置を示す。 プロトロンビン分子は単一のポリペプチド鎖からなり、そしてプロトロンビンの部分タンパク質分解によって形成されたトロンビンは単一のジスルフィド結合によって一緒に結合した2つのポリペプチド鎖からなる。
血液(真皮および単球)との接触は、一定の条件下でのみ起こります:血管の損傷および/またはそれらの原形質膜の通常の非対称性の破壊の場合。 タンパク質分解活性化における組織因子は必要ありません。
第V因子と第VIII因子 -ドメインタンパク質が血中を循環している。 第V因子は肝臓で合成され、第VIII因子は内皮細胞によって合成されます。 両因子はトロンビンによる部分的タンパク質分解によって活性化される。 血漿中の第VIII因子はvon Willebrand's protein - platelet factorと組み合わされます。 この複合体中のフォンビルブラント因子は第VIII因子を安定化させ、抗凝固相の前抗分解酵素による第Ca因子によるその破壊を防止する。
酵素複合体と細胞膜との相互作用イオンCa 2+の関与により生じる。 凝固促進性経路の全ての利益(II、VII、IX、X)は、肝細胞のERにおけるこれらのタンパク質の翻訳後修飾から生じるu-カルボキシグルタミン酸残基を含む。
第VIIa因子、第IXa因子および第Xa因子中のγ-カルボキシグルタミン酸残基は、Ca 2+を介して細胞膜の負に荷電したリン脂質とこれらの酵素との相互作用を確実にする。 Ca 2+イオンの不在下では、血液は凝固しない。
凝血促進剤凝固経路のプロ酵素におけるグルタミン酸残基のカルボキシル化におけるビタミンKの役割 凝血促進経路の前駆体中のグルタミン酸残基のカルボキシル化はカルボキシラーゼによって触媒され、その補酵素はビタミンK(ナフトキノン) - ビタミンKジヒドロキノンの還元型である(セクション3を参照)。
摂取されたビタミンK(ナフトキノン)は、ビタミンKジヒドロキノンの形成を伴うNADPH依存性ビタミンKレダクターゼによって肝臓で回復されます。 K.エポキシドのビタミンKジヒドロキノンへの再生は以下のように起こる:第1に、その補酵素がタンパク質であるチオール依存性エポキシドレダクターゼによってビタミンKの2,3−エポキシドがビタミンKに回復する、p。 チオレドキシンは承認した。 その後、この反応で生成されたビタミンKは、ビタミンKジヒドロキノン中のチオール依存性レダクターゼを持つ酵素ビタミンKによって修復されます。

図 14−13。 グルタミン酸の翻訳後カルボキシル化におけるビタミンKの役割 1 - 外因性ビタミンK NADPH依存性レダクターゼの回収。 因子II、VII、IX、X、プロテインC、ビタミンK依存性カルボキシラーゼ中のグルタミン酸残基の2-γ-カルボキシル化は、2,3-エポキシドビタミンKの形成を伴うジヒドロキノンの酸化を伴う。 3- 2,3-エポキシドチオール依存性ビタミンKレダクターゼの回収。 4 - ビタミンKチオール依存性ビタミンKレダクターゼの回収。 a)およびb) - チオレドキシン様タンパク質の還元型および酸化型。
この反応は、前の反応と同様に、チオレドキシン様タンパク質です(図14-13)。 ビタミンK欠乏症は、凝固促進経路の機能のカルボキシル化障害につながり、出血、皮下および内出血を伴います。
ビタミンK構造類似体であるジクマロールとワルファリンは、チオール依存性酵素であるビタミンK 2,3-エポキシドレダクターゼとビタミンKレダクターゼを阻害し、血液凝固を阻害します(図14-14)。 これらの薬は血栓症を予防するために臨床現場で使用されています。
凝固促進経路反応のカスケードの開始。凝固促進性経路の酵素膜複合体は、原形質膜の外表面に組織因子細胞および負に帯電したリン脂質が存在する場合にのみ形成される。 特に、原形質膜の横方向の非対称性は、外層における中性リン脂質(ホスファチジルコリンおよびスフィンゴミエリン)、および内層における負に帯電した(ホスファチジルイノシトールビスホスフェートおよびホスファチジルセリン)の優位性によって決定される(セクション5参照)。 特別な酵素体系は、細胞の原形質膜の外側表面が通常荷電していない、細胞膜内の膜貫通移動およびリン脂質の分布を提供する(セクション5参照)。
血小板膜および内皮細胞の横方向の非対称性が乱されると、それらの表面に負に帯電した(血栓形成性)領域が形成され、そして組織因子アポタンパク質IIIが露出される。 このような違反は怪我をすることで発生する可能性があります。 この場合、組織因子および細胞膜の内面は、凝固促進経路の血漿因子に利用可能になる。 さらに、血栓形成を引き起こすシグナル伝達分子と内皮細胞の受容体および血小板との相互作用は、Ca 2+依存性調節システムを活性化する(セクション5を参照)。 最終的に、これは細胞質中のCa 2+の含有量の増加をもたらし、それはATP依存性アミノリン脂質トランスロカーゼを阻害する。 この酵素はリン酸 - チジルセリンを外側の脂質層から内側の脂質層に転移させるので、膜の横方向の非対称性を保存するのに重要な役割を果たす。 アミノリン脂質トランスロカーゼの活性の低下は、細胞膜の外層におけるホスファチジルセリンの含有量の増加および膜酵素複合体の形成に必要な負に帯電した領域の形成をもたらす。 さらに、このような原形質膜構造の破壊の結果として、組織因子がその外面に露出され、そして凝固促進性凝固経路VII-TF-Ca 2+の最初の酵素複合体が形成される。

各複合体の酵素の活性化は、その全成分の相互作用の結果です。 第IX因子、第X因子および第II因子が活性化を必要とする場合、第VII因子は低いタンパク質分解活性を有する。 部分的タンパク質分解による膜複合体VII-TF-Ca 2+の第VII因子は、第IXおよびX因子を活性化する。活性因子IXaおよびXaは、膜複合体IXa-VIIIa-Ca 2+およびXa-Va-Ca 2+の形成に関与する。 同時に、第Xa因子は第V因子をタンパク質分解的に活性化し、そしてプロトロンビナーゼ複合体はプロトロンビンをトロンビンに変えるだけでなく、複合体VIIa − Tf − Ca 2+におけるタンパク質分解活性が複合体VII − Tf−におけるより10,000倍高い第VII因子も活性化する。 Ca 2+。
反応のカスケードの結果として形成されたトロンビンは、フィブリノーゲンの部分タンパク質分解、第XIII因子の反応を触媒し、そして正のフィードバックの原理により、第V、VIIおよびVIII因子をタンパク質分解的に活性化する。
凝固過程では、シグナル増幅には2つのメカニズムがあります。各酵素結合がシグナル増幅をもたらす一連の反応と、ポジティブフィードバックです。
B.血液凝固接触経路
血液凝固の接触経路は、損傷第XII因子と損傷した内皮表面との相互作用から始まる
図 14−15。 血液凝固の凝固促進(外部)および接触(内部)経路の図。 名称:IUD - 高分子量キニノーゲン。 TF - 組織因子。 → - 血液凝固因子の活性化 ・・・\u003e正のフィードバックに基づく凝固因子の活性化 酵素複合体のβ - 膜リン脂質成分。 血液凝固膜複合体のすべての酵素はプロテアーゼであり、そして部分的タンパク質分解によって活性化される。 亜内皮との接触の結果として活性化された1-第XII因子はプレカリクレインをカリクレインに変える。 2 - カリクレイン膜複合体カリクレイン-IUDは第XII因子を活性化する。 3−第XIIa因子は第XI因子を活性化する。 部分的タンパク質分解によって活性化された4因子XIIaは、正のフィードバックの原理に従ってプレカリクレインをカリクレインに変換する。 5−第XIa因子膜複合体XIa − BMKは第IX因子を活性化する。 6−第Xa因子(Xa膜複合体IXa − VIIIa − Ca 2+は第X因子を活性化する; 7,8−第VIIa因子複合体VIIa − Tf − Ca 2+は第IX因子およびX因子を活性化する; 9−プロトロンビナーゼ複合体のXa因子は第II因子を活性化する; 10) 1,11-トロンビン(第II因子)は、フィブリノーゲンをフィブリンに変換し、そして第XIII因子を活性化する; 12-第XIIIa因子は、フィブリンゲルにおけるアミド結合の形成を触媒する。
血管壁。 この相互作用は第XII因子の活性化をもたらしそして凝固の接触相の膜酵素複合体の形成を開始する。 それらは、酵素カリクレイン、第XIa因子(トロンボプラスチンの血漿前駆体)および第XIIa因子(Hageman因子)、ならびに活性化タンパク質 - 高分子量キニノーゲン(VMC)を含む(図14-15)。
第XII因子- 血の中で循環している職業。 それは2つの方法で逐次的に活性化される:第一に、損傷を受けた内皮の負に荷電した表面と相互作用するときの立体構造の変化の結果として、次にカリクレイン−VMC膜複合体との部分タンパク質分解によって。
高分子キニノーゲン -酵素膜複合体XIIa-IUD、Xla-BMKおよびカリクレイン-IUDにおけるタンパク質活性化因子。 VMCは、肝臓で合成され、120kDの分子量を有する血漿糖タンパク質である。 それは、血液凝固の接触相のタンパク質分解酵素とコラーゲン内皮下との相互作用を媒介し、そしてさらに、カリク - レイン - キニン系の成分である。
カリクレイン- 基質が、第XII因子に加えて、血漿血漿タンパク質プラスミノーゲン(フィブリンの溶解に関与するプロ酵素)および低(69kD)および高(120kD)分子量を有するキニノーゲンであるセリンプロテアーゼ。 キニノーゲンの部分的タンパク質分解は調節性キニンペプチドを産生する。 特に、強力な血管拡張薬ブラジキニンは血管透過性を高め、内皮細胞膜の破壊を引き起こします。
第XII因子と血管内皮下組織との接触の結果として、それは活性化される。 タンパク質分解的にIUDと複合体を形成する活性因子XIIa
iUDを通して膜に関連したプレカリクレインをカリクレインに変える。 部分タンパク質分解による正のフィードバックの原理に基づくカリクレイン-IUD膜複合体は第XII因子を活性化する。 同時に、第XII因子は最大の酵素活性を獲得し、そして正のフィードバックの原理により、IUDと関連したプレメタリックレインを活性化する。 さらに、部分的タンパク質分解の結果として形成された第XIIa因子は第XI因子をタンパク質分解的に活性化し、そして酵素複合体XIa-IUDの組成中の第XI因子は第IX因子を活性化する。 1Xa - VIIIa - Ca 2+膜複合体の第IXA因子は、プロトロンビナーゼ複合体内でプロトロンビンを活性化する第X因子を活性化する。
トロンビンの形成をもたらす一連の反応は、2つの方法、すなわち凝固促進剤(外部)および接触(内部)で実現することができる(図14〜15)。 外部経路の反応を開始するためには、血液と接触している細胞の原形質膜の外側表面上の組織因子の出現が必要である。 内部経路は、血管内皮の損傷表面との接触による第XII因子の活性化、およびプレカリクレイン酵素と第XII因子の相互活性化から始まる。
かくして、血液凝固の凝固促進および接触経路において、膜酵素複合体の逐次形成は第X因子の活性化およびプロトロンビナーゼの形成をもたらす。 血液凝固の両方の方法について同じである段階は、一般的な血液凝固と呼ばれます。 現在、UPA-TF-Ca 2+複合体は第X因子よりも第IX因子をより効果的に活性化し、第VII因子は第1Xa因子によって活性化されるが明らかに遅いので、内部および外部凝固経路の概念はかなり条件付きと考えられる。 対活性化因子Xa。 したがって、血液凝固反応のカスケードは、2つの比較的独立した経路に沿ってではなく、主に直線的な順序で起こると仮定することができる。 接触経路は明らかに凝固を開始するために絶対に必要というわけではない。 明らかに、それは血液凝固系を身体の様々な調節系、例えば、カリクレイン - キニンおよび血栓を溶解する線維素溶解酵素系と結び付けるのに役立つ。
健康な人の血 in vitro5〜10分で凝固します。 この場合、プロトロンビナーゼ複合体の形成には5〜8分かかり、プロトロンビンの活性化には2〜5秒かかり、フィブリノーゲンからフィブリンへの変換には2〜5秒かかります。
血液凝固を減らしました。血液凝固性が低下すると、繰り返し出血を伴う疾患が観察されます。 血友病は出血の増加を特徴とする遺伝性疾患です。 これらの出血の原因(自発的または外傷によって引き起こされる)は、血液凝固系タンパク質の遺伝的な不足です。
血友病A(古典的血友病)は、X染色体上に局在する第VIII因子遺伝子の突然変異によって引き起こされます。 古典的血友病は血友病の全症例の80%を占める。 血友病Bはあまり一般的ではなく、第IX因子の遺伝的欠陥によって引き起こされます。
第VIII因子遺伝子の欠損は劣性形質として現れ、それゆえ男性だけがこの形の血友病にかかっています。 この疾患は皮下、筋肉内および関節内の出血を伴い、時に生命を脅かす。 第VIII因子の欠損は、10,000人の新生児のうち約1人に発生します。 患者は、ドナー血液から得られた、または遺伝子工学的方法によって得られた、第VIII因子を含む調製物で治療される。
G.血液抗凝固薬システム
血液凝固の生理学的阻害剤は、それらが血液を液体状態に保ちそして血管の損傷領域の外側への凝血塊の広がりを防止するので、止血を維持するのに重要な役割を果たす。
凝固促進剤と血液凝固の接触経路との反応の結果として形成されるトロンビンは、血栓からの血流によって洗い流される。 それは、血液凝固酵素のインヒビターと相互作用することによって不活性化されるか、または血栓の形成を阻害する抗凝固相を活性化することができる。
抗凝固相血液凝固は空間だけでなく時間的にも制限されるべきです。 抗凝固相
血中の活性因子の寿命を制限し、トロンビン自体によって開始されます。 その結果、トロンビンは、一方では血液凝固を促進し、凝固反応のカスケードにおける最後の酵素であり、他方ではそれを減速させ、無傷の血管内皮上に抗凝固相の酵素複合体を形成させる。 この段階は短いカスケードの反応であり、そこではトロンビン、タンパク質活性化因子トロンボモジュリン(Tm)、ビタミンK依存性セリンプロテアーゼタンパク質C、タンパク質活性化因子Sならびに第Va因子および第VIIIa因子が関与する(図14-16)。
抗凝固相の一連の反応において、2つの膜複合体IIa − Tm − Ca 2+およびCa − S − Ca 2+が順次形成される。
トロンボモジュリン- 内皮細胞膜の不可欠なタンパク質。 それはタンパク質分解活性化を必要とせずそしてトロンビンの活性化剤として役立つ。 トロンビンはトロンボモジュリンと相互作用した後に初めてプロテインCを活性化する能力を獲得し、そしてトロンボモジュリンと結合したトロンビンはフィブリノーゲンをフィブリンに変換することができず、第V因子および血小板を活性化しない。
プロテインC- γ-カルボキシグルタメート残基を含有する利益。 膜複合体IIa-Tm-Ca 2+中のトロンビンは、部分的タンパク質分解によってプロテインCを活性化する活性化プロテインC(Ca)は、膜結合型活性化因子タンパク質Sを形成する

図 14−16。 抗凝固相 Tm - トロンボモジュリン。 C - プロテインC; Ca活性タンパク質C; S - プロテインS; 脂肪線 - 膜結合複合体。 1−トロンビン(Na)は、タンパク質トロンボモジュリン(Tm)と膜複合体を形成する。 2-膜複合体Ia - Tm - Ca 2+の組成中のトロンビンはプロテインCを活性化する。 Ca - S - Ca 2+酵素膜複合体の組成中の3-活性化プロテインCは、第Va因子および第VIIIa因子中の2つのペプチド結合を加水分解し、それらを不活性ペプチドに変える。
複雑なCa-S-Ca 2+。 この複合体の一部としてのCaは、第Va因子および第VIIIa因子中の2つのペプチド結合を加水分解し、そしてこれらの因子を不活性化する。 3分間錯体Ca − S − Ca 2+の作用下で。 第VIIIa因子と第Va因子の活性の80%が失われています。 したがって、トロンビンは、正のフィードバックの原理により、その形成を促進するだけでなく、プロテインCを活性化することによっても血液凝固過程を阻害する。
プロテインCおよびSの遺伝的欠損は、第VIIIa因子および第Va因子の不活性化速度の低下をもたらし、そしてそれは血栓性疾患を伴う。 プロテインCに耐性のある第V因子を合成する第V因子遺伝子の突然変異もまた血栓形成をもたらす。
抗凝固相は血液凝固カスケードの阻害を引き起こし、そして凝固酵素阻害剤は血流中の活性酵素を不活性化する。
血液凝固酵素の阻害剤血液凝固酵素の生理学的阻害剤は血管への損傷部位への血栓の広がりを制限する。 血漿タンパク質 アンチトロンビンIII- 血液凝固の最も強力な阻害剤。 それは、血液抗凝固活性の約80〜90%を占めます。 トロンビン、第IXa因子、第Xa因子、第XIIa因子、カリクレイン、プラスミンおよびウロキナーゼといった一連の血中セリンプロテアーゼを不活性化します。 アンチトロンビンIIIは、第VIIIa因子を阻害せず、そして膜複合体の組成中の因子に影響を及ぼさないが、血漿中にある酵素を排除し、血流中の血栓形成の拡大を防ぐ。
アンチトロンビンと血液凝固酵素との相互作用は、ヘパリンの存在下で促進されます。 ヘパリンは肥満細胞で合成されるヘテロ多糖類です。 ヘパリンとの相互作用の結果として、アンチトロンビンIIIは、血液のセリンプロテアーゼに対するその親和性が増大する立体構造を獲得する。 アンチトロンビンIII-ヘパリン - 酵素複合体の形成後、ヘパリンはそれから放出されそして他のアンチトロンビン分子と結合することができる。
若年時のアンチトロンビンIIIの遺伝性欠乏症では、生命に危険な血栓症および血管塞栓症が観察されます。
α2 - マクログロブリン血液のセリンプロテアーゼと複合体を形成します。 そのような複合体では、それらの活性中心は完全には遮断されていません、
そしてそれらは小さな基質と相互作用することができる。 しかしながら、フィブリノーゲンのような高分子基質は、α2 - マクログロブリントロンビン複合体中のプロテアーゼの作用にアクセスすることができなくなる。
抗転換(外部凝固経路の組織抑制剤)血管内皮で合成されます。 それはTf-VIIa-Ca 2+酵素複合体に特異的に結合し、その後それは肝臓により捕捉されそしてその中で破壊される。
α1 アンチトリプシントロンビン、第XIa因子、カリクレインを阻害するが、血液凝固因子の重要な阻害剤とはみなされず、α1 - アンチトリプシンは膵臓および白血球のプロテアーゼ、コラゲナーゼ、レニン、およびウロキナーゼを組織レベルで阻害する。
職業およびプロファクターのタンパク質分解活性化の結果として形成されたペプチドもまた顕著な抗凝固特性を有するが、それらの作用のメカニズムはまだ明らかではない。
D.止血における血小板の役割
血小板が損傷した血管壁表面に接着(付着)し、そして互いに結合(凝集)し、血小板血栓を形成し、そして血管の損傷部位に止血因子を分泌する能力が、止血におけるそれらの役割を決定する。
血中を循環する血小板は円盤形をしており、血管の無傷の内皮には付着しません。 付着および凝集は、血小板および無傷の内皮、ならびにプロスタサイクリン(PG 12)の相互反発を妨げる。 いくつかのインデューサーの作用機序および血小板凝集のリプレッサーは、図1において考察されている。 14〜17。
プロスタサイクリン血管内皮のアラキドン酸から形成され、血流に入ります(セクション8を参照)。 内皮細胞によるプロスタサイクリンの合成および分泌は、トロンビン、ヒスタミン、アンジオテンシンIIおよびカリクレインを刺激する。 それはアデニル酸シクラーゼシグナル伝達系を通してその作用を実現する(第5節参照)。 プロスタサイクリンと受容体との相互作用は、プロテインキナーゼAの活性化を引き起こす。活性型プロテインキナーゼAはリン酸化し、したがってCa 2+ -ATP-アズおよびCa 2+トランスロカーゼを活性化する。 これは、血小板の細胞質におけるCa 2+のレベルの低下、それらの円盤状の形状の保存および凝集能力の低下をもたらす。
血小板の活性化は、ホスファチジルセリンによって形成された負に帯電した領域の原形質膜表面上の出現を伴う。
血小板活性化および凝集の主な誘発物質はフォンビルブランド因子、コラーゲン、トロンビン、ADPである。
ファクター・フォン・ヴィレブランド -血漿、血管内皮および血小板α顆粒に存在する糖タンパク質。 血管壁が損傷した場合、コラーゲン、基底膜および内皮下筋細胞はフォンビルブランド因子を介して血小板と相互作用する。 血小板の原形質膜はこの因子に対する数種類の受容体を含む。 von Willebrand因子は、受容体と相互作用して、イノシトールリン酸シグナル伝達系を介して血小板に作用します(第5節参照)。 最終的に、これは血小板の細胞質中のCa 2+含有量の増加および複合体カルモジュリン-4Ca 2+ - ミオシンキナーゼの形成をもたらす。 この複合体の組成中の酵素ミオシンキナーゼは収縮性タンパク質ミオシンをリン酸化し、それはアクチンと相互作用してアクトミオシン(トロンボステニン)を形成する。 結果として、血小板はスパイク球状の形状を獲得し、それらが互いにそして損傷を受けた内皮の表面とそれらの相互作用を促進する。
フォン・ヴィレブランド因子の濃度を下げる、その数を減らす、またはその構造を変えると、癒着や血小板の凝集が悪くなり、それに伴って出血が起こります。 これは、血小板中のフォンビルブラント因子受容体糖タンパク質Iaの欠如によるバーナード - ソリエ症候群、およびフォンビルブラント因子の欠乏によるフォンビルブラント病において観察される。
血小板活性化の最も重要な主な誘発物質はトロンビンとコラーゲンです。 これらのタンパク質と血小板の原形質膜の特異的受容体との相互作用は、密集した尿細管系から細胞質へのCa 2+の移動をもたらす。
最終的にそれらの付着と凝集を引き起こす。
コラーゲンホスホリパーゼA2の血小板活性化を引き起こし、それはそれらの膜のリン脂質からアラキドン酸を放出する。 アラキドン酸は酵素シクロオキシゲナーゼ(COX)の基質として働く。 シクロオキシゲナーゼにより触媒される反応の結果として、環状エンドペレキスプロスタグランジンG 2(PG G 2)およびプロスタグランジンH 2(PG H 2)が形成される。 これらのプロスタグランジンは、トロンボカンシンセターゼの作用によってトロンボキサンA 2に変換される(第8節参照)。 トロンボキサンA 2はcAMPのレベルを低下させ、そしてホスホリパーゼCを活性化することにより、密な管状系からのCa 2+の放出を促進する(図14〜17)。
トロンビン7つの膜貫通ドメインを持つ不可欠なタンパク質である、特定の受容体と相互作用します。 トロンビンは、部分的タンパク質分解によってレセプターを活性化し、そこから血小板の外側血漿表面に位置するN末端ペプチドを切断する。 それ故、トロンビンは、他の活性化剤とは異なり、触媒的に作用し、そして1分子のトロンビンはいくつかの受容体を活性化し得る。 信号が送信されます
![]()
結果として生じるカルモジュリン-4Ca 2+ミオシンキナーゼの複合体はミオシンをリン酸化し、そのアクチンとの相互作用は血小板の形態の変化、それらの接着および凝集をもたらす。 さらに、プロテインキナーゼCは血小板タンパク質プレクストリンをリン酸化する。 ホスホリル化プレクトリンは、顆粒に含まれる血小板活性化および凝集の血小板含有二次誘導物質に対して「放出反応」を引き起こす。 これらの物質には、ADP、Ca 2+、GDF、セロトニン、ヒスタミン、および小顆粒中に存在するβ-トロンボグロブリンタンパク質、フォンビルブラント因子、フィブロネクチンタンパク質、トロンボスポジンおよびIUDが含まれ、これらは血小板の高密度顆粒に含まれる。 トロンボス - ポジンは血小板同士の相互作用に関与しています。 p-トロンボグロブリンはプロスタサイクリンの分泌を減らし、ヘパリンに結合します。 フィブロネクチンはコラーゲン、ヘパリンおよび血小板に対する結合部位を有する。
ADP 血小板に含まれており、また赤血球が破壊されると血流に入ります。 ADPは特定の受容体と相互作用し、アデニル酸シクラーゼ活性を阻害します。 これは細胞内Ca 2+の動員の増加を引き起こし、そして最終的に血小板凝集をもたらす。
したがって、血小板の活性化はそれらの代謝の変化および生物学的に活性な物質の放出を伴う。 これらの物質は、形態変化、癒着、血小板凝集を引き起こし、血栓の形成に関与しています。
受容体の機能的活性および血小板の二次メディエーターの系の破壊はそれらの機能の変化をもたらしそして血栓症または出血を含む多くの疾患の原因となり得る。
血小板凝集を侵害する薬は血栓症の発生を予防するために使用されます。 アスピリン(シクロオキシゲナーゼ阻害剤)、ニコチン酸(トロンボキサン合成酵素阻害剤)およびCa 2+ブロッカーは血小板凝集を阻害し、血栓形成シグナルの異なる段階に影響を及ぼす。
E.線維素溶解
血栓は形成後数日以内に溶解する。 線維素溶解は、血管床から除去される可溶性ペプチドを形成するためのフィブリン繊維の酵素的分解である。 血栓の組成中のフィブリンの破壊は、セリンプロテアーゼプラスミンの作用下で起こる。
プラスミン活性化剤の作用によってプラスミノーゲンから形成される。 プラスミンプラスミノーゲンの不活性なプロ酵素は、肝臓、腎臓および骨髄で合成されます。
組織プラスミノーゲンアクチベーター(TAL) -肝臓を除くすべての組織の血管内皮に含まれるタンパク質分解酵素。 血中のこの活性化物質の放出は、感情的なストレス、痛み、静脈血栓塞栓症、中等度の身体活動で増加します。 TALは、部分タンパク質分解によって不活性プラスミノーゲンを活性プラスミンに変換する。 プラスミノーゲンアクチベーターはまた、第XIIa因子およびカリクレインである。
フィブリンクロットの溶解は、フィブリン、プラスミノーゲンおよびTAPの相互作用中に起こる(図14-18)。
血栓の形成中のフィブリン繊維の網状構造の形成は、プラスミノーゲンおよびその活性剤のその上への収着を伴う。 プラスミンおよびプラスミノーゲンの分子には、フィブリンドメインに相補的な領域があり、プラスミン1分子はいくつかのフィブリン分子に結合することができる。 TAP分子はフィブリン結合部位も有する。 TAPプラスミンによって形成されたプラスミノーゲンは、フィブリンを加水分解して、線維素溶解を活性化するペプチドXおよびY、ならびにそれを阻害するペプチドDおよびEを形成する。 可溶性ペプチドX、Y、D、Eは血流に入り、そこで貪食される。 血栓の破壊はそれからのプラスミンおよびTAPの放出をもたらす。 血流中では、後者は特定の阻害剤によって急速に不活性化され、そして肝臓によって捕捉される。
TAPは、第一(i - TAP - 1)および第二(および-TAP - 2)型の組織活性化剤プラスミンの阻害剤によって、およびα2 - 抗プラスミンまたは他のセリンプロテアーゼ阻害剤によってプラスミンが阻害される。
腎臓では、タンパク質分解性プラスミノーゲンアクチベーターであるウロキナーゼが合成され、これがプラスミノーゲンをプラスミンに変換して、

図 14−18。 線維素溶解のスキーム 1-活性化剤(第XIIa因子、カリクレイン、TAP)の作用下でフィブリン塊に吸収されたプラスミノーゲンは、部分タンパク質分解によってプラスミンに変換される。 2−プラスミンはフィブリンを加水分解して可溶性ペプチドX、Y、D、Eを形成する。 3 - 血流中で、TAPは特定のタンパク質i-TAP-1およびi-TAP-2によって不活性化される。 4-プラスミン活性は、セリンプロテアーゼの非特異的阻害剤(α2 - 抗プラスミン、α2 - マクログロブリン、α1 - アナ - トリプシン、アンチトロンビン - ヘパリン複合体)の作用下で低下する。
フィブリン繊維からの腎臓糸球体の放出。
β溶血性連鎖球菌から、プラスミノーゲンと複合体を形成するストレプトキナーゼタンパク質が単離され、ここでプラスミノーゲンは自己触媒的にプラスミンに変換される。
ウロキナーゼ、ストレプトキナーゼおよびTAPは、心筋梗塞、静脈血栓症および動脈血栓症の血栓溶解療法、ならびに血液透析において使用されている。
α2 - マクログロブリン、α1 - アンチトリプシンおよびアンチトロンビンIII - ヘパリン複合体のような血液凝固酵素のそのような阻害剤もまた、小さな線維素溶解活性を有する。
血液の線溶活性の低下は血栓症を伴う。 フィブリンクロットの破壊は、遺伝性プラスミン遺伝子欠損またはその構造の遺伝的欠陥、血中へのプラスミノーゲンアクチベーターの流れの減少、ならびに血中のフィブリン溶解阻害剤の含有量の増加(i-TAP-1および-TAP-2、α2 - アンチプラスミン)によって引き起こされ得る。
遺伝性および後天性の止血障害は、出血を特徴とする出血性疾患と血栓性疾患の両方を引き起こす可能性があります。 しかしながら、血栓症および血管内凝固(血栓増加症)への傾向の増加は血友病よりはるかに一般的であることに注意すべきです。 例えば、血友病は、国によって異なりますが、10万人あたり6〜18人の間で、抗トロンビンIIIの欠乏によって引き起こされる血栓症は、1〜2人の患者で5,000人、プロテインCがない場合は15,000人で発生します。 男の人
血液凝固 多細胞生物の細胞は生きており、それら自身の液体培地と接触する。 この媒体は血漿、組織液、リンパ液からなり、体液の内部環境と呼ばれます。 構成においては、それは生物全体を取り巻く外部環境とは異なります。 それ故、その自然な過程の中でこの液体内部環境の保全におけるその完全性の違反の場合には極めて重要な必要性がある。 高等脊椎動物およびヒトでは、進化の過程で血液凝固系が出現しました。 さらに、高等生物における凝固システムの価値は、血管壁の完全性に違反して止血または出血を止めるという概念よりもはるかに広い。
血液凝固は体の保護反応です。 血管から放出された血液は3〜4分以内に凝固する、すなわちそれは液体からゲル様状態になる。 血液凝固は、血漿フィブリノーゲンの可溶性タンパク質が不溶性フィブリンに変換されるという事実による。
血液凝固はいくつかの段階で起こります。 第一段階、すなわち一次止血、または前段階は、それがそうであるように、第二段階に先行して開始します - 実際には凝固、それは今度は多相過程です。 その本質は化学的酵素反応から成り、その結果として血液凝固因子に活性物質が現れます。
一次止血
これはいくつかの段階で起こる複雑な生理学的過程です。 その主な参加者は血管壁、神経系および血小板です。 原発性止血は主に反射性の原発性血管攣縮から始まります。 その後、いわゆる内皮血小板反応が始まります。 損傷部位では、血管の内皮がその電荷を変える。 血管内の辺縁位置を占める血小板は、血管の損傷表面に付着(付着)し始め、それらの間で凝集(一緒に付着)する。 結果として、2〜3分後、第3段階、すなわち「血小板ネイル」の形成段階が始まります。 この段階で出血は止まりますが、血液凝固はまだ起きていません。 血漿は液体のままです。 結果として生じる血栓はゆるく、そして短時間の間、プロセスは可逆的である。 第4段階は、結果として生じる血栓において、血小板の形態学的変換が始まり、それがそれらの不可逆的な変化および破壊につながることである。 これは粘性の血小板変態です。 粘性変態の結果として、その中に含まれる凝固因子は血小板から出てくる。 それらの相互作用は微量のトロンビンの出現をもたらし、それが一連の化学的酵素反応 - 酵素的凝固を引き起こす。
酵素凝固
微量のトロンビンの出現は、いわゆる酵素的凝固の複雑な過程を引き起こす。
酵素的凝固の第一段階は、血液と組織凝固因子との多段階の相互作用の結果として、以前に欠けていた因子であるトロンボプラスチンが血液中に現れると始まる。 第二段階はトロンボプラスチンとプロトロンビン、トロンビンの不活性前駆体との相互作用です。 カルシウム塩の存在下でのトロンボプラスチンとプロトロンビンの相互作用の結果として、活性トロンビンは凝固段階が始まるのに十分な濃度で血中に現れる - トロンビンと可溶性フィブリノーゲンとの相互作用および後者の不溶性フィブリンへの移行。 これが第3段階です。 診療所における最初のフィブリンフィラメントの出現に従って、血液凝固時間が決定される。
従って、血液の酵素的凝固プロセスは3段階で進行する:1−活性トロンボプラスチンの形成、2−活性トロンビンの発生および3−不溶性フィブリンフィラメントの沈殿。
それから、次の酵素段階が始まります。その間に、血栓の凝縮と収縮が起こります。これは、凝固能力を失った透明な液体血清の分離です。 これは、血液凝固の第4段階 - 血栓の収縮(圧迫)です。 そして最後に、最後の5番目の段階が始まります - 血栓の溶解(溶解)。 それはまた、多くの物質の酵素的相互作用が起こる多段階プロセスであり、最終的には活性酵素 - フィブリノリシンの出現をもたらす。 フィブリノリジンはフィブリンの糸の間の結合を破壊し、それを再び不溶性フィブリノーゲンに翻訳する。 現在、体の独立した線溶系の存在について話すのが慣例です。 もちろん、体内のこれらのプロセスははるかに複雑であり、さらに多くの要因が関係しています。
回転血液系 (同義: 凝固システム、止血システム、血液凝固) - 血管の完全性および血液の液体状態を維持しながら、フィブリン血栓の形成を通して出血の停止をもたらす酵素系。 S.p. に - 機能部品フィジオル。 血の凝集状態の調節のシステム(見なさい)。
血液凝固の理論の基礎(参考文献)はA. A. Schmidtによって開発されました。 彼は酵素反応の結果として血液凝固の最初の段階でカットに従って、二相血液凝固の理論を定式化しました、トロンビンフィブリノーゲン(参照)の影響の下で第2段階でフィブリンになります(参照)。 1904年、Moravitz(R.O. Morawitz)、次にSalibi(V.S.Salibi、1952)およびOvren(PA Owren、1954)は、血漿中のトロンボプラスチンの形成を発見し、プロトロンビンの変換におけるカルシウムイオンの役割を示した。 トロンビン これは、血液凝固の三相理論を公式化することを可能にし、それに従ってプロセスが連続的に進行する:第一相において活性プロトロンビナーゼが形成され、第二に - トロンビンの形成、第三に - フィブリンの出現。
McFarlenスキームによれば、血液凝固はカスケード型で進行する、すなわち不活性因子(プロファメント)は徐々に活性酵素に変換され、それが次の因子を活性化する。 したがって、血液凝固は、フィードバックの原理に基づいて作用する複雑な多段階のメカニズムです。 このような変換の過程において、その後の変換の速度および活性化物質の量は増加する。
酵素的連鎖反応である血液の凝固は、血液凝固因子と呼ばれる血漿、血小板、および組織の成分を含みます(止血を参照)。 血漿(凝固促進剤)、組織(血管)および細胞性(血小板、赤血球など)の血液凝固因子があります。
主な血漿因子は、第I因子(フィブリノゲンを参照)、第II因子(プロトロンビンを参照)、または組織トロンボプラスチン、第IV因子、またはイオン化カルシウム、第VII因子、またはコラー因子(Proconvertinを参照)、第V因子、第X因子です。 、XI、XII、XIII(出血性素因を参照)、第VIII因子およびIX因子(血友病を参照)。 第III因子(血栓形成因子) - 体内のすべての組織に見られるリン脂質タンパク質。 第VII因子およびカルシウムと相互作用すると、それは第X因子を活性化する複合体を形成する。第II因子、第V因子(Ac-グロブリン)、第VII因子、第IX因子、第X因子、第XI因子、第XII因子および第XIII因子は酵素である。 第VIII因子(抗血友病グロブリン - AGH)は凝固酵素の強力な促進剤であり、第I因子と共にそれは非酵素群を構成する。
組織因子、カリクレイン - キニン酵素系の成分(キニンを参照)は、血液凝固および線維素溶解の活性化に関与している:血漿プレカリクレイン(Fletcher因子、XIV因子)および高分子キニノーゲン(Fitzgerald因子、Williams因子、Flogger因子、XV因子)。 組織因子には、血管内皮で合成されたフォンビルブラント因子、線維素溶解の活性化剤および阻害剤(参考文献)、血小板凝集の阻害剤であるプロスタサイクリン、ならびに第XII因子および血小板接着を活性化する亜内皮構造(参考文献)が含まれる 。
凝固性血小板因子群は細胞性血液因子と呼ばれ、そのうちリン脂質(膜)血小板因子3(3tf)およびタンパク質抗ヘパリン因子(因子4)、ならびにトロンボキサンAr(プロスタグランジンG2)、赤血球細胞が重要である。 血小板第3因子の類似体(エリスロプラスチン、エリスロサイクチン)など
条件付きでは、血液凝固機構は、外部(組織から血液への組織トロンボプラスチンの進入によって引き起こされる)および内部(血液または血漿に含まれる酵素因子によって引き起こされる)、または第X因子の活性化段階までのライ麦、またはスチュアートプラウエラ因子に分けられ得る。 そしてプロトロンビナーゼ複合体の形成は、種々の凝固因子の関与を伴ってある程度別々に行われ、そしてその後共通の経路に沿って実施される。 血液凝固のカスケード複雑なメカニズムは、図に示されています。
2つの血液凝固メカニズムには複雑な関係があります。 かくして、外部機構の影響下で、血小板凝集を刺激し、血小板因子を放出し、第VIII因子および第V因子を活性化するのに十分な少量のトロンビンが形成され、それが第X因子のさらなる活性化を促進する。 トロンビン中の第Xa因子およびプロトロンビン。 血液凝固メカニズムにおける第XII因子の一見重要な役割にもかかわらず、それが欠乏している場合には出血はなく、血液凝固時間の延長のみが起こる。 おそらくこれは、コラーゲンと組み合わせた血小板が第XII因子の関与なしに第IX因子と第XI因子を同時に活性化する能力によるものであろう。
カリクレイン - キニン系の成分は血液凝固の初期段階の活性化に関与しており、第XII因子は興奮剤です。 カリクレインは第XI因子1aと第XI因子の相互作用に関与し、第VII因子の活性化を促進する、すなわちそれは血液凝固の内的および外的メカニズムの間の関連として作用する。 第XV因子も第XI因子の活性化に関与しています。 血液凝固の異なる段階で、複合体タンパク質 - リン脂質複合体が形成される。
地殻内では、カスケード方式の時間が変わり、追加が行われます。
内部機構による血液の凝固は、過剰なカテコールアミン(例、アドレナリン)、プロテアーゼが血流中に現れると、コラーゲンおよび結合組織の他の成分(血管壁が損傷している場合)と接触している第XII因子(接触因子またはHageman因子)の活性化で始まる。 また、血液や血漿が体外の異物表面(針、ガラス)と接触するためです。 同時に、その活性型が形成されます - HNa因子は、血小板の3因子と一緒に形成されます。これは、XI因子の酵素として作用するリン脂質(3 TF)で、それをX1a型の活性型に変えます。 カルシウムイオンはこの過程に関与しません。
第IX因子の活性化はそれに対する第X1a因子の酵素効果の結果であり、カルシウムイオンは第1Xa因子の形成に必要である。 第VIII因子(ヴィラ因子)の活性化は第1Xa因子の影響下で起こる。 第X因子の活性化は、カルシウムイオンの存在下における第IXa因子、第VIIa因子および第3TF因子の複合体によって引き起こされる。
血液凝固の外部メカニズムにおいて、組織および臓器から血液中への組織トロンボプラスチンは第VII因子を活性化し、それと組み合わせてカルシウムイオンの存在下で第X因子の活性化剤を形成する。
内部および外部メカニズムの一般的な経路は、比較的安定したタンパク質分解酵素である第X因子の活性化から始まります。 第X因子の活性化は、それが第Va因子と相互作用すると1000倍加速される。 第Xa因子と第Va因子、カルシウムイオンおよび3tfとの相互作用によって形成されたプロトロンビナーゼ複合体は、第II因子(プロトロンビン)の活性化をもたらし、その結果トロンビンが形成される。
血液凝固の最終段階は、フィブリノーゲンの安定化フィブリンへの変換である。 タンパク質分解酵素であるトロンビンは、最初に2つのペプチドA、次に2つのペプチドBのα鎖とβ鎖から切断します。その結果、4つの遊離結合を持つフィブリンモノマーが存在します。 次に、トロンビンによって活性化された第XIII因子(フィブリン安定化因子)の関与により、安定化された、または不溶性のフィブリンが形成される。 フィブリン塊は多くの赤血球、白血球および血小板を含み、それらもまたその固化をもたらす。
従って、全てのタンパク質凝固因子が酵素であるとは限らず、従って他のタンパク質の分解及び活性化を引き起こすことができないことが確立されている。 血液凝固の異なる段階で、酵素が活性化され、そして非酵素成分がこの活性化を加速および増強し、そして基質に対する作用の特異性を提供する因子の複合体が形成されることも確立された。 このことから、カスケード方式はカスケード複合体と見なすべきであることがわかります。 それは様々な血漿因子の相互作用の順序を保存するが、その後の段階に関与する因子を活性化する複合体の形成を提供する。
血液凝固系でもいわゆる区別されます。 止血の血管血小板(一次)および凝固(二次)メカニズム(参照)。 血管血小板機構が観察されるとき、血小板の塊による損傷血管の閉塞、すなわち細胞止血栓の形成。 このメカニズムは、低血圧の小血管にかなり信頼できる止血をもたらします。 血管壁が損傷している場合は、けいれんがあります。 露出したコラーゲンおよび基底膜は創傷表面への血小板付着を引き起こす。 続いて、フォンビルブラント因子の関与による血管障害の領域における血小板凝集および凝集が起こり、血小板凝固因子の放出が起こり、血小板凝集の第二相、二次血管痙攣、フィブリン形成が起こる。 フィブリン安定化因子は高級フィブリンの形成に関与している。 血小板血栓の形成における重要な役割は、カルシウムイオンの存在下での群れの影響下で、ADPに属し、血小板は互いにくっついて凝集体を形成する。 ADPの供給源は血管壁、赤血球および血小板壁のATPである。
凝固メカニズムでは、主な役割はページのS.の要因に属します。 v。両方とも通常コンジュゲートとして機能するので、血管血小板の単離および止血の凝固機構は相対的である。 外傷性因子への曝露後に出血が発生する時点までに、その原因を推定することが可能である。 血漿因子に欠陥があると、それは血小板減少症よりも遅く起こります(参照)。
体内では、血液凝固のメカニズムと一緒に、循環血液の液体状態をサポートするメカニズムがあります。 B. A. Kudryashovの理論によると、この機能はいわゆるによって実行されます。 抗凝固剤系では、切断の主なリンクは酵素的および非酵素的線維素溶解であり、これは血流中の血液の液体状態を提供する。 他の研究者(例えばA. A. Markosyan、1972)は、抗凝固メカニズムを単一の凝固システムの一部と考えています。 Sの相互関係が確立されます。 なぜなら、線維素溶解系だけでなく、キニン(参考文献)および補体系(参考文献)も同様だからです。 活性化第XII因子はそれらの引き金です。 さらに、それは第VII因子の活性化を促進します。 3. S。Barkagan(1975)および他の研究者によると、結果として、第XII因子が機能し始めます - 血液凝固と線維素溶解の内部と外部のメカニズムの間のカリクレイン「橋」は同時に活性化されます。 抗凝固システム(抗凝固システム)は反射性を持っています。 それは血流中の化学受容体の刺激時に比較的過剰のトロンビンの血流中の出現により活性化される。 そのエフェクター作用は、組織供給源からのヘパリン(参照)および線維素溶解活性化因子の血流への放出を特徴とする。 ヘパリンは、アンチトロンビンIII、トロンビン、フィブリノーゲン、および他の多くの血栓形成タンパク質、ならびにカテコールアミンと複合体を形成する。 これらの複合体は抗凝固活性を有し、安定化されていないフィブリンを溶解し、非酵素的にフィブリンモノマーの重合を遮断し、そして第XIII因子のアンタゴニストである。 酵素的線維素溶解の活性化により、安定化した血餅の溶解が起こる。
タンパク質分解酵素阻害剤の複雑な系は、プラスミン、トロンビン、カリシレイン、および活性化凝固因子の活性を阻害する。 それらの作用のメカニズムは、酵素と阻害剤との間のタンパク質 - タンパク質複合体の形成に関連している。 7つの阻害剤が見出された:α−マクログロブリン、インター−I−トリプシン阻害剤、C1−不活性化剤、α− 1−抗キモトリプシン、アンチトロンビンIII、α− 2−アンチプラスミン、α−抗トリプシン。 ヘパリンは即時の抗凝固作用があります。 トロンビンの主な阻害剤はアンチトロンビンIIIであり、これはトロンビンの75%、ならびに他の活性化凝固因子(1Xa、Xa、CPA)およびカリクレインを結合する。 ヘパリンの存在下では、アンチトロンビンIII活性は劇的に増加します。 血液のアンチトロンビン電位の25%を提供し、カリクレインの活性を完全に阻害するA2” MacRグロブリンは、血液凝固にとって重要である。しかし、カリクレインの主な阻害剤は、第XII因子を阻害するC1阻害剤である。フィブリンも抗トロンビン効果を有する。 トロンビンによりフィブリノーゲンから切断されるフィブリンおよびフィブリノペプチドに対する抗ポリメラーゼ効果を有するフィブリン/フィブリノーゲンのタンパク質分解生成物(S.s.k.の活性の破壊は酵素プラスミンの高い活性を引き起こす(Fiber参照)。 noliz)。
体内の凝固因子は、止血を確実にするのに必要な量よりもはるかに多く含まれています。 しかしながら、抗凝固剤が存在するので血液は凝固せず、そして止血の過程において、少量の凝固因子、例えばプロトロンビンのみが、血液凝固の自己遅延および神経内分泌調節機構のために消費される。
S.の違反。 を基にすることができます。 臨床的に血管血栓症(血栓症を参照)、出血性素因(参考文献を参照)、ならびに血の凝集状態の調節システムにおける関連障害、例えば血栓性出血症候群(を参照)、またはマチャベリ症候群の形で現れる過程。 止血の変化は、さまざまな血小板異常、血管、血漿凝固因子、またはこれらの組み合わせに起因する可能性があります。 違反は、定量的および/または定性的、すなわち、何らかの要因の欠乏または過剰、その活性または構造の破壊、ならびに血管、器官および組織の壁の変化に関連し得る。 それらは後天的(毒性化合物の影響、感染、電離放射線、タンパク質損傷、脂質代謝、癌、溶血)、遺伝性または先天性(遺伝的欠陥)です。 取得した違反の中で、S. pの逸脱につながっています。 〜、最も頻度が高いのは、低機能性貧血における骨髄機能の抑制、napr、またはVerlgof病における過剰な血小板の破壊、napr(紫斑血小板減少症)です。 後天性および遺伝性の血小板変性症もしばしば遭遇する(参照)。ライムギは、血小板の殻の質的欠陥(例えば、膜糖タンパク質の欠乏)、および血小板放出の反応の結果である。 血小板凝固因子などの含有量
凝固因子の欠如または特異的抗体によるそれらの阻害のために、出血が増加する可能性があります。 多くの血液凝固因子が肝臓で形成されるので、第II因子、第V因子、第VII因子、第IX因子、第X因子、または肝性ジ(低)フィブリノゲン血症の濃度の低下により出血が頻繁に起こる。 場合によっては出血を伴うK-ビタミン依存性因子(II、VII、IX、X)の欠乏が、腸への胆汁の流れ(閉塞性黄疸)、ビタミンK拮抗薬(クマリン、ワルファリン)の過剰摂取、腸内細菌叢、および出血性疾患に違反して観察される 新生児(出血性素因を参照)。
とのS.の活性化の結果として。 特に、組織トロンボプラスチン(手術、重度の傷害、火傷、ショック、敗血症など)は、しばしば完全で不完全な播種性血管内凝固症候群を発症します(血栓出血症候群を参照)。 S.指標 に
主要フィジオールの遺伝性または後天性の欠乏は、播種性血液凝固および血栓症の発症に寄与する。 抗凝固薬、特に抗トロンビンIII、および線維素溶解系の成分。 輸血補充療法を必要とするこれらの物質の二次的な消耗は、血液凝固の過程およびアンチトロンビンIIIの代謝を増加させるヘパリンの集中的使用の両方におけるそれらの集中的消費の結果であり得る。 。
血管壁における脂質代謝の侵害および炎症過程は、血管壁における構造的変化、その内腔の有機的狭窄をもたらし、これは血餅の形成(例えば、心筋梗塞において)の引き金として役立ち得る。 血栓形成因子を含む赤血球の過剰な破壊もまた、例えば発作性夜間血色素尿症および自己免疫性溶血性貧血(溶血性貧血を参照)、鎌状赤血球貧血の間に、血栓の形成のための必要条件であることが多い(参考文献)。
ほとんどの場合、凝固因子の欠乏は遺伝的に決定されています。 したがって、血友病患者には第VIII、第IX、第XI因子の欠乏が観察されます(参考文献)。 出血の増加は、第X因子、第XIII因子および低フィブリノゲン血症または無フィブリノゲン血症と同様に、第II因子、第V因子、第VII因子の欠乏(低プロコンベルティン血症を参照)に起因する。
血小板の遺伝的機能的劣性は、例えば、血小板凝集の障害および血栓の収縮を特徴とする、大群の疾患、例えばGlantsmann's血栓無力症の根底にある(血小板減少症を参照)。 血小板顆粒の成分の放出の反応障害またはADPおよび他の凝集刺激剤の血小板における蓄積障害(いわゆる蓄積プール病)を伴う出血性素因が記載されている。 多くの場合、血小板減少症と合併した血小板減少症(Bernard病 - Soulierなど)。 血小板凝集の障害、顆粒の欠損、ADPの含有量の減少が、Chediak-Higashi異常で見られました(血小板減少症を参照)。 血小板機能不全の原因は、血小板の接着および凝集の過程に関与する血漿タンパク質の欠乏であり得る。 したがって、ウィレブランド因子欠乏症の場合、内皮下および外表面への血小板の接着が破壊され、同時に第VIII因子の凝固活性が低下し、その成分の一つがウィレブランド因子である。 フォンビルブラント - ユルゲンス病(血管血友病を参照)では、これらの障害に加えて、血小板のリン脂質因子3の活性が低下します。
研究方法S.p. 〜は出血、血栓症および血栓出血の原因を突き止めるために使用されます。 凝血する能力は一連の方法によって調査され、それは異なる条件下での凝血塊の出現率の決定である。 おおよその値を持つ最も一般的な方法は、血液凝固時間(参考文献)、出血時間(参考文献)、血漿再石灰化時間および抗凝固療法をモニターするために使用されるOvreneトロンボテストの確立です。 血漿再石灰化時間を決定する際に、蒸留水および塩化カルシウム溶液を検査すべき血漿に添加する。 血栓形成時間を固定する(時間を長くすると出血する傾向があり、凝固亢進が短くなることを示す)。 Ovrenトロンボテストでは、検査対象の血漿に試薬が添加されます。試薬には、II、VII、IX、X因子を除くすべての血液凝固因子が含まれています。 血漿凝固の遅延はこれらの因子の欠乏を示す。
より正確な方法には、ヘパリンに対する血漿耐性を決定するために使用されるZigg法、トロンボエラストグラフィー(参照)、トロンビン時間(トロンビン参照)およびプロトロンビン時間を決定する方法(参照)、トロンボプラスチン生成試験、またはトロンボプラスチン形成のBiggs法が含まれる。 カオリン - ケファリノボゴ時間の決定法Douglas Biggs-Douglasトロンボプラスチン形成法では、アルミナ水和物で治療された健康な人の血漿と血小板が研究対象の血清に加えられます。 この場合の血漿凝固遅延は凝固因子の欠乏を示す。 カオリン - ケファリン時間を決定するために、血小板が乏しいカオリンの懸濁液および塩化カルシウムの溶液を血漿に添加する。 血漿凝固の時点までに、第VIII因子、第IX因子、第XI因子および第XII因子の欠乏ならびに過剰の抗凝固剤が確立され得る。
血液の線維素溶解活性はユーグロビン、gistokhyによって決定されます。 方法など(線維素溶解を参照)。 追加の方法、例えば、第XII因子と第VII因子との間のカリクレイン架橋の低温活性化を検出するための試験、パラ凝固の生成物、生理学的抗凝固剤、抗血栓形成活性、フィブリノーゲン分解の生成物などを決定する方法がある。
参考文献: Fib − rhinolysis、M.、1979、bibliogr。 B.Alu − dおよびV.P.など。止血システム研究の実験室的方法、Tomsk、1980年。 S.出血性疾患および症候群、M。 動物と人間の生化学、エド。 MDクルスク他、c。 6、s。 3、94、Kiev、1982年。 O. Gavrilov。血液の凝集状態の調節システムの生物学的規則性とそれらを研究する仕事、Probl。 ヘマトール。 と輸血、24巻、7号、p。 3,1979; 急性放射線症の出血性症候群、ed。 T.K.Dzharykyana、J.I。、1976、bibliogr。 血友病とその治療、編。 D.Fedorova、L.、1977年、参考文献; 4。 Georgieva S.A.およびKl I hkおよびn JI。 M.血液凝固および線維素溶解に対する薬物の副作用、Saratov、1979年、参考文献; J。 A. I.医薬品と血液凝固、Kiev、1978年。 Kudryashov B. - 血液の液体状態の調節とその凝固の生物学的問題、M。、1975、参考文献; I。およびSkipetrov V.P.に造られた。血液、血管壁、止血および血栓形成要素、M、1974年。 Marcosian A.A.血液凝固の生理学、M。 M and-chabelis MS With Agulopathic syndrome、M.、1970; Mについてsh。G.心血管疾患における血栓症および塞栓症、それを伴う経路。 ルーマニア人から。ブカレスト、1979年。 血液凝固系の個体発生、ed。 ; A.A.Markosian、L.、1968年、参考文献; A。 血液凝固の理論における問題点と仮説、ed。 O.K.Gavrilova、M.、1981、bibliogr。 Rabi K.ローカライズされ、散在したvirusgso-sudny凝固、trans。 、1974年、M。 M.と3 akとD. D.のzh。Antitromboticheskaya療法、Baku、1979年:Savel'ev V.S、Ya bkについてE. G.とToとr I.肺動脈の血栓塞栓症、M。 Skipetrov V.P.とK.Z.Z.とB. B. B. II。 産科血栓出血性症候群、イルクーツク - ■チタ、1973年。 ubおよびubおよびM.小児血液学、トランス。 英語では、M。 Filatov A.N.およびKotovschinaM.A.A。臨床診療における血液凝固システム、L.、1963年、参考文献; J。 A.およびTithova M.I.心臓、血管および肺の外科的疾患における止血のシステム、M。 1977年; Chazov E.I.およびLakin K.M. 血液凝固と止血、エド。 J.M.トムソン著、エジンバラ - N. 止血、生化学、生理学および病理学、ed。 D. Ogston著a。 B.Bennett、L。 止血および血栓症、ed。 G. G. Neri Serneriによるa。 C.R.Prentice、L.a。 、1979年:ヒト血液凝固、止血および血栓症、ed。 R. Biggs著、オックスフォード、1976年。 Nilsson I. M.出血性および血栓性疾患、L.a。 、1974年。 化学的線維素溶解および血栓溶解における進歩、ed。 1978年、J.F。 Quick A.J.出血性疾患および止血の病理学、スプリングフィールド、1974年。 血友病の最近の進歩、ed。 1975年、L.M.AledortによるN. 静脈および動脈血栓症、病因、診断、治療、編 J.H. Joistによるa。 L. A Sherman、N. Y.、1979。
OK K. Gavrilov。
関連記事